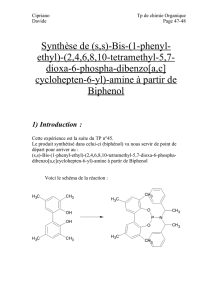
Réactions de fluoration de dérivés azotés insaturés en milieu

THESE
Pour l’obtention du Grade de
DOCTEUR DE L’UNIVERSITE DE POITIERS
Faculté des Sciences Fondamentales et Appliquées
(Diplôme National-Arrêté du 7 août 2006)
Ecole Doctorale : Sciences pour l’Environnement Gay Lussac.
Secteur de recherche : Chimie organique, Minérale et Industrielle
Présentée par :
Fei LIU
***************************
REACTIONS DE FLUORATION DE DERIVES AZOTES
INSATURES EN MILIEU SUPERACIDE
***************************
Directeurs de thèse :
Marie-Paule JOUANNETEAUD, Professeur, Université de Poitiers
Sébastien THIBAUDEAU, Maître de Conférences, Université de Poitiers
***************************
Soutenue le 02 juillet 2010
devant la commission d’examen
***************************
JURY
M. T. LEQUEUX
Professeur, ENSICAEN, Caen
Rapporteurs
M. T. BRIGAUD
Professeur, Université de Cergy-Pontoise
M. R. GREE
Directeur de recherche, Université de Rennes1
Examinateurs
M. F. ZUNINO
Directeur de société @rtMolécule
Mme M-P. JOUANNETEAUD
Professeur, Université de Poitiers
M. S. THIBAUDEAU
Maître de Conférences, Université de Poitiers
Mme A. MINGOT
Maître de Conférences, Université de Poitiers

1
SOMMAIRE ......................................................................................................... 1
INTRODUCTION ................................................................................................ 5
GENERALITES ................................................................................................... 6
I. Propriétés du fluor et des composés fluorés ..................................................................... 7
I.1 Effets structuraux ....................................................................................................... 7
I.2 Propriétés physiques ................................................................................................... 8
- I.2.1 Point d'ébullition ...................................................................................... 8
- I.2.2 Polarité ...................................................................................................... 8
- I.2.3 Lipophilie ................................................................................................. 8
I.3 Propriétés électroniques et réactivité ......................................................................... 9
- I.3.1 Force de liaison ......................................................................................... 9
- I.3.2 Distribution électronique ....................................................................... 10
- I.3.3 Interactions dipôle C-F/charge ............................................................. 10
- I.3.4 Acidité et Basicité ................................................................................... 11
- I.3.5 Stabilité des intermédiaires réactionnels .............................................. 11
II. Dérivés azotés fluorés en chimie médicinale ................................................................. 13
II.1 Augmentation de la stabilité métabolique .............................................................. 13
II.2 Les peptides mimétiques fluorés : inhibition d’enzymes ........................................ 14
II.3 Modification de l’affinité enzymatique : interaction fluor-protéine ...................... 14
II.4 Diminution de la basicité : effet sur l’affinité drogue-récepteur ............................ 15
III. Synthèse de fluoroamines ............................................................................................ 17
III.1 Synthèse de fluoroamines par voie nucléophile..................................................... 17
- III.1.1 Substitution nucléophile d’aminoalcool .............................................. 17
- III.1.2 Utilisation du DFBA (N, N-diéthyl-
,
-difluorobenzylamine) .......... 18
- III.1.3 Ouverture d’aziridines......................................................................... 19
- III.1.4 Substitution d’halogènoamines ........................................................... 20
- III.1.5 Fluoroalkylation nucléophile de sulfinilimines ................................... 20
III.2 Synthèse de fluoroamines par voie électrophile .................................................... 21
- III.2.1
-fluoration d’imines et réduction ...................................................... 21
- III.2.2 Aminofluoration palladocatalysée ....................................................... 22
III.3 Synthèse multi-étapes de fluoroamines : utilisation de synthons fluorés ............. 22

2
- III.3.1 Utilisation d’alcools fluorés ................................................................. 22
- III.3.2 Utilisation de dérivés fluorohalogénés ................................................. 23
III.4 Synthèse de fluoroamines par réaction en milieu superacide ............................... 24
IV. Superacides ................................................................................................................... 26
IV.1 Définition ................................................................................................................ 26
IV.2 Types de superacides .............................................................................................. 26
IV.3 Milieu HF/SbF5 ....................................................................................................... 27
IV.4 Carbocations ........................................................................................................... 27
IV.5 Réactions en milieu superacide .............................................................................. 28
- IV.5.1 Isomérisation ....................................................................................... 28
- IV.5.2 Polymérisation ...................................................................................... 29
- IV.5.3 Réarrangements et cyclisations .......................................................... 29
- IV.5.4 Halogénation ........................................................................................ 30
IV.6 Réactions de fluoration en milieu superacide ........................................................ 31
V. Superélectrophile ........................................................................................................... 34
V.1 Différentes classes de superélectrophiles................................................................. 34
- V.1.1 Superélectrophiles gitoniques................................................................ 34
- V.1.2 Superélectrophiles distoniques ......................................................... 35
V.2 Superélectrophiles provenant de dérivés azotés ................................................... 36
- V.2.1 Dications iminium-carbénium ............................................................... 36
- V.2.2 Dications ammonium-carbénium ......................................................... 38
- V.2.3 Réactivité des dications ammonium-carbénium .................................. 40
RESULTATS et MECANISMES ...................................................................... 42
I. Réaction d’hydrofluoration ............................................................................................ 43
I.1 Rappel sur la réactivité des amines insaturées en milieu superacide ...................... 44
I.2 Extension de la réaction d’hydrofluoration d’amines insaturées ............................ 46
- I.2.1 Influence de la substitution sur l’azote................................................... 46
- I.2.2 Influence de la substitution de la double liaison .................................... 52
- I.2.3 Réaction d’homodimérisation/fluoration ............................................... 58
II. Réactivité de sulfonamides aromatiques....................................................................... 64
II.1 Intérêt et synthèse de sulfonamides en chimie médicinale ..................................... 65
- II.1.1 Propriétés biologiques des sulfonamides .............................................. 65

3
- II.1.2 Propriétés biologiques des sulfonamides cycliquesErreur ! Signet non défini.68
- II.1.3 Synthèse de sulfonamides cycliques ...................................................... 68
II.2 Cyclisation et fluoration de N-allyles benzènesulfonamides dans HF/SbF5 .......... 71
- II.2.1 Rappel sur les sulfonamides obtenus en milieu superacide au
laboratoire ................................................................................................................... 71
- II.2.2 Mise au point des conditions opératoires en milieu superacide
HF/SbF5 ............................................................................... Erreur ! Signet non défini.72
- II.2.3 Influence de la substitution sur l’aromatique....................................... 74
- II.2.4 Application à la synthèse de sultams originaux .................................... 78
- II.2.5 Synthèse de 4-aminobenzène sulfonamides cycliques ou fluorés ......... 87
III. Synthèse de dérivés azotés gem-chlorofluorés et gem-difluorés ................................. 91
III.1 Rappel des résultats obtenus au laboratoire ......................................................... 92
III.2 Réaction des amines allyles chlorées...................................................................... 93
- III.2.1 Résultats ............................................................................................... 94
- III.2.2 Formation des amines gem-chlorofluorés et gem-difluorés ................ 94
III.3 Influence de la substitution sur l’azote et de la distance entre l’azote et la
double liaison .................................................................................................................. 96
- III.3.1 Résultats ............................................................................................... 96
- III.3.2 Détermination des structures .............................................................. 98
- III.3.3 Mécanisme et discussion .....................................................................101
III.4 Réactivité des imides insaturés en milieu superacide ..........................................103
- III.4.1 Résultats ..............................................................................................103
- III.4.2 Détermination des structures .............................................................106
- III.4.3 Mécanisme et discussion .....................................................................109
- III.4.4 Utilisation des nouveaux dérivés azotés gem-chlorofluorés ...............118
CONCLUSION................................................................................................. 120
PARTIE EXPÉRIMENTALE ......................................................................... 123
I. Manipulation de HF et de SbF5 .....................................................................................124
II. Réactions en milieu HF/SbF5 .......................................................................................124
III. Suivi des réactions et purification ..............................................................................124
IV. Analyse des produits ...................................................................................................124

4
V. Réaction hydrofluoration d’amines insaturées ............................................................125
VI. Synthèse de Sulfonamides ...........................................................................................132
VII. Synthèse de sultams originaux ..................................................................................143
VIII. Synthèse de 4-aminobenzène sulfonamides cycliques ou fluorées ..........................149
IX. Synthèse de composés gem-chlorofluorés ...................................................................152
X. Synthèse de composés gem-difluorés ............................................................................156
XI. Modélisation et RMN in situ des intermédiaires réactionnels ...................................163
XII. Utilisation des nouveaux dérivés azotés gem-chlorofluorés .....................................169
BIBLIOGRAPHIE ........................................................................................... 172
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
1
/
181
100%