Que faire devant une hyperferritinémie?

135
••••••••
hyperferritinémie:a)Une lysecellu-
laire,hépatiqueou musculaire; b)Une
induction de la synthèsedeferritine
par l’alcool; c)Une dérégulation de la
synthèsedeferritine du fait de muta-
tions dans le gène de laferritine.
Comment,en pratique,
conduirelediagnostic
d’une hyperferritinémie?
Nepas méconnaître
une hyperferritinémie modérée
Du fait de l’étenduedela zone de nor-
malitéindiquée par les laboratoires
d’analyse,avecdes limites supérieures
de lanormale de l’ordrede 300-
400 µg/lchez l’homme et de 200-300
chez lafemme,il y a risquede sous-
estimer une hyperferritinémie réelle
lorsqueles chiffres avoisinent le ver-
sant haut de cettefourchette. Ainsi,
chez lafemme,la valeur normale est
de l’ordrede 30 µg/lavant laméno-
pauseet monteenmoyenne graduel-
lement vers 80 après laménopause.
C’est direqu’un taux de 200 peut
correspondre,chez lafemme,à une hy-
perferritinémie tout àfait significative.
La rapporter
à une hémochromatose
de type 1(ou hémochromatose
liée au gène HFE)[3]
Enl’absencedecontextecliniquenet-
tement évocateur,c’est l’élévation
conjointedu taux de saturation de la
Quefairedevant
une hyperferritinémie?
transferrine quiconstituel’élément
d’orientation déterminant.Étant donné
quedans l’hémochromatose,l’éléva-
tion de laferritine sérique signifie déjà
constitution d’unexcès tissulaireen
fer,le taux de saturation de la trans-
ferrine sera toujours nettement élevé,
le plus souvent ≥80%(N<45%).
La confirmation du diagnostic
d’hémochromatosede type 1 repose
dès lors sur lapositivitédu test géné-
tique:mutation C282Yprésenteàl’état
homozygotequipermet d’affirmer
l’hémochromatoseliée au gène HFE.
Une fois le diagnosticposé,le dosage
de laferritinémie intervient dans la
priseencharge de l’hémochromatose
àplusieurs niveaux ainsiqu’il aété
récemment précisédans les Recom-
mandations de laHauteAutoritéde
Santé(HAS)[4]:a)Quantification de la
surcharge en fer:en effet,le degré
d’élévation de laferritine constitue,en
cette situation, unexcellent reflet du
degréd’excès en fer (en l’absencede
facteurs associés susceptibles d’inter-
férer dans cetteaugmentation, tels
qu’unalcoolisme et/ou un syndrome
polymétabolique) :il y aen effet dans
l’hémochromatose une très bonne
Qu’est-cequelaferritine
et quels sont les
principaux mécanismes
d’une hyperferritinémie?
[1, 2]
Ledosage de laferritine sériqueest un
examen biologique très demandé pour
juger du statut martial. Ilpermet en
effet,àlafois de renseigner sur l’exis-
tenced’undéficit en fer (hypoferriti-
némie) et sur celle d’une surcharge (hy-
perferritinémie). L’interprétation d’une
augmentation du taux sériquedefer-
ritine dépasse toutefois le seulcadre
des excès en fer et supposedonc une
analysepertinentedelapart du mé-
decin prescripteur.
La ferritine est une protéine ressem-
blant à une coquille d’oeufcapable de
stocker du fer en son sein (jusqu’à4500
atomes de fer par molécule de ferri-
tine),cequienfait laprotéine de
stockage du fer par excellence,en par-
ticulier au niveau des hépatocytes et du
système macrophagique(hépato-
splénique). Mais laferritine est égale-
ment une protéine de la réaction
inflammatoire, saproduction aug-
mentant en situation d’activation
macrophagique.
Ces deux fonctions, stockage
tissulairedu fer et expression de
l’inflammation, situent les deux
domainesphysiopathologiques prin-
cipaux auxquels peut être rapportée
une hyperferritinémie. Trois autres
mécanismes peuvent expliquer une
P.BRISSOT,
Caroline LE LAN,
Marie-BérengèreTROADEC,
Anne GUILLYGOMARC’H,
R.LORHO,
Anne-Marie JOUANOLLE,
D.GUYADER,
R.MOIRAND, O.LOREAL,
Y.DEUGNIER (Rennes)
Tirés àpart :PierreBrissot,Servicedes Maladies du Foie,HôpitalPontchaillou - 35033
Rennes.

de lanormale (300 µg/Lchez l’homme;
200 chez lafemme) puis toutes les deux
saignées ensuitejusqu’àobtention de
ladésaturation (ferritine<50µg/L). En
période d’entretien,le rythme de suivi
conseillé est uncontrôle de ferritinémie
toutes les deux saignées.
Dans cecadreHFE, unprofil d’hété-
rozygotie composite(C282Y/H63D)
peut parfois être repéré. Engénéral,
les taux d’hyperferritinémie et d’aug-
mentation de la saturation de la trans-
ferrine sont alors modérés (à titrein-
dicatif respectivement inférieurs à500
et 65%) saufco-facteurs d’élévation
de ces paramètres tels quel’alcoolisme
et/ou le dysmétabolisme. Ilimporte
notamment de rappeler qu’une hyper-
ferritinémie,quel qu’en soit le niveau,
ne peut être rapportée à une hétéro-
zygotie composite,et doncà une forme
marginale d’hémochromatoseHFE, si
la saturation de la transferrine est
normale (<45%).
La rapporter à un syndrome
dysmétabolique[7]
L’hyperferritinémie dysmétabolique(ou
«hépatosidérosedysmétabolique»)
constitueaujourd’huileplus fréquent
diagnosticdifférentiel de l’hémochro-
matose:de nombreux diagnostics er-
ronés d’hémochromatose sont en pra-
tiqueportés devant, typiquement, une
franche hyperferritinémie (de l’ordre
de 600-1200)quiconduit souvent le
médecin àdemander à tort (c’est-à-
dire sans avoir préalablement contrôlé
le taux de saturation de la transfer-
rine) un test génétiqueHFE, lequel
repèrepar exemple une simple hété-
rozygotie C282Y(voire une hétérozy-
gotie H63D),ces résultats génétiques
étant considérés à tort comme étayant
le diagnosticd’hémochromatose... En
outre,il n’est pas rarequ’uncontrôle
échographiquehépatique soit effectué
et que saconclusion libellée «foie de
surcharge » soit considérée par le
médecin non spécialistecomme un
argument diagnostiquedeplus,alors
quec’est d’une surcharge en graisse
dont il s’agit (l’excès en fer ne pou-
vant jamais êtredétectépar uncontrôle
échographique).
Enfait,il est essentiel de rappeler que:
a)le test génétiquenedoit êtrede-
mandé qu’après s’êtreassuréqu’il y a
bien élévation de la saturation de la
transferrine (≥45%et en particulier
≥60%chezl’homme et ≥50%chez
lafemme); b)quel’hémochromatose
HFE ne peut être rendue responsable
d’hyperferritinémie sans élévation
conjointedela saturation de la trans-
ferrine (la seule exception est repré-
sentée par lacoexistencefortuited’un
syndrome inflammatoiremarquéqui
non seulement majorele taux de fer-
ritine mais diminue voirenormalisele
taux de fer sériqueet celuidela satu-
ration de la transferrine) ; c)qu’un
simple état d’hétérozygotie C282Y(et
afortioriH63D)nepeut jamais être
rendu responsable d’une élévation
significativedelaferritinémie. La seule
réserveest représentée par d’excep-
tionnels cas rapportés d’hétérozygotie
compositedont lamutation associée
àC282Yn’est pas H63D(donnant donc
l’impression,au vu du résultat du test
génétiquede routine,d’une hétéro-
zygotie C282Y«simplex»); mais,en
pareil cas,la saturation de la trans-
ferrine est franchement élevée,cequi
situed’emblée dans uncadrequine
peut être une simple hétérozygotie
C282Y.
Cettehyperferritinémie dysmétabo-
lique sedémarquedel’hyperferriti-
némie hémochromatosiquepar les
corrélation entreferritinémie et concen-
tration hépatiqueenfer;b)Evaluation
pronostique:le chiffrede1000 cor-
respond à un seuil critiqueen terme de
toxicité viscérale. Enparticulier,au
planhépatique,il aétébien montré
quelorsquele taux de ferritinémie est
inférieureà1000 et quene s’y asso-
cient ni hépatomégalie ni augmentation
du taux d’ASAT, point n’est besoin de
réaliser une ponction-biopsie hépatique
car il n’y aalors pas de risquedefibrose
significative[5]; c)Valeur décision-
nelle au plan thérapeutiquepuisque
c’est àpartir du stade2 de laclassifi-
cation de l’expression phénotypiquede
l’homozygotie hémochromatosique[4,
6](caractérisépar l’élévation conjointe
des taux de saturation de la transfer-
rine et de laferritine sans signes cli-
niques associés)qu’est posée l’indica-
tion de lamiseen routedu traitement
déplétif; d) Valeur en tant quepara-
mètrede suividel’efficacitédu traite-
ment.Ainsi, tant en phased’induction
qu’en phased’entretien des saignées,
c’est l’appréciation du taux de ferriti-
némie quiguide le schéma thérapeu-
tiquedont l’objectif globalest l’obten-
tion puis le maintien d’un taux
<50µg/L.Concernant le rythme de sur-
veillancedelaferritinémie,l’HAS
recommande qu’en phased’induction,
il soit bimestriel tant quele taux n’apas
encoreatteint les limites supérieures
136
••••••••
Ferritine
= Cytolyse
=Hémochromatose
Sat. Tf ?
=Inflammation
Dysmétabolisme
Acéruloplasminémie
Gaucher
Cataracte
Mutation Ferroportine
Transa. ?
N
CRP ?
N
N
±
++
0
Fer
hépatique
hépatique
musculaire
HFE
non HFE
(IRM)
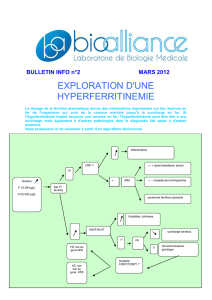
Figure1.–Diagnostic schématiqued’une hyperferritinémie. (Sat.Tf=Saturation
de laTransferrine ; Transa.= transaminases ; CRP =CRéactiveProtéine ;
=augmentation ; N= valeur normale)
➡

comptedel’éventuelle dominante
d’élévation de l’ASAT par rapport à
l’ALAT.
–Le syndrome inflammatoiregénéral:
l’hyperferritinémie y est habituelle-
ment modérée (< 500 µg/L)et s’ac-
compagne d’une hyposidérémie (et
d’une baissedela saturation de la
transferrine). Importancepar consé-
quent de vérifier le taux de CRP
devant toutehyperferritinémie.
–L’alcoolisme chronique[8]: une hy-
perferritinémie (parfois >1000 µg/L)
est observable chez l’alcooliqueen
l’absencede toutecytolyseet de sur-
charge en fer (du fait d’une stimu-
lation de synthèsedelaferritine par
l’alcool),associée dans lamoitié des
cas à une hypersidérémie. Après
sevrage,le fer senormaliseenmoins
d’une semaine alors queladécrois-
sancedelaferritine est plus lente
pour se stabiliser après trois mois
d’abstinence. Enpratique,il faut
donc réfréner lapratiquedu taux
de ferritine en situation d’alcoolisme
«actif»aigu et/ou chronique sous
peine de «construire» nombre
de faux diagnostics d’hémochro-
matose...
La rapporter àd’autres causes
rares ou exceptionnelles
Certaines de ces causes s’accom-
pagnent d’une surcharge viscérale en
fer,les autres non.
SITUATIONS RARES
OU EXCEPTIONNELLES
D’HYPERFERRITINÉMIE
AVEC SURCHARGE EN FER
Elles peuvent êtreclassées selon le taux
de saturation de la transferrine plas-
matique.
Surcharges génétiques en fer avecélé-
vation de la saturation de la transfer-
rine :le taux de saturation de la trans-
ferrine y est le plus souvent de l’ordre
de 100%. Lephénotype est donc très
proche de celuidel’hémochromatose
liée àHFE, en sorteque seuls ces
syndromes devraient pouvoir être
intitulés «hémochromatoses»non liées
àHFE [9].
Chez le sujet de moins de 30 ans:
évoquer une hémochromatosejuvé-
nile (ou hémochromatosede type 2).
éléments suivants :a)la saturation de
la transferrine est normale; b)l’excès
hépatiqueenfer,le plus souvent dé-
sormais évaluépar l’IRM hépatique,
n’est en règle quemodéré,inférieur à
2 à 3 fois lalimite supérieurede
lanormale en terme de concentration
hépatiqueenfer,c’est-à-dire
<120 µmol/gdefoie secpour une
N<36.Ilexisteainsidans ce syndrome,
une disproportion entrel’assez haut
niveau de ferritinémie (quipeut
dépasser 1000 µg/L)et le degrémo-
dérédel’hépatosidérose,cequi sou-
lèveleproblème du mécanisme de cette
hyperferritinémie quipourrait plus
relever d’une activation macropha-
giquequed’une surcharge en fer;c)il
existebien sûr un terrain polyméta-
bolique,associant peu ou prou une
surcharge pondérale, une hyper-
lipidémie, unDNID, une HTA, une hy-
peruricémie (voire une goutte).
Il vade soi quelorsqu’une hémochro-
matoseauthentique s’associe à un
syndrome dysmétabolique(et/ou à
unalcoolisme), son profil biologique
peut s’en trouver modifié avec une
hyperferritinémie plus importanteque
ne le voudrait le degréd’excès en fer.
C’est en cettecirconstancequela réa-
lisation d’une IRM hépatiqueest jus-
tifiée afin d’éviter d’être«piégé»dans
l’estimation du degré réel de surcharge
hépatiqueenfer par unniveau d’hy-
perferritinémie quin’est alors plus le
seul reflet de laconcentration hépa-
tiqueenfer.
La rapporter
àd’autres causes fréquentes
–La cytolyse:a)d’origine hépatique:
qu’il s’agissed’une hépatiteaiguë
(surtout)ou chronique,l’hyperfer-
ritinémie peut alors s’accompagner
d’uncertain degréd’hypersidérémie
et d’élévation de la saturation de la
transferrine (surtout s’il y ainsuf-
fisancehépatocellulaireassociée),
d’où l’importancedecoupler au
dosage de laferritine le contrôle des
transaminases (ALATet ASAT); b)
d’origine musculaire:myolyse
cardiaqueou périphérique(rhabdo-
myolyse),d’où l’utilitéd’uncontrôle
conjoint des enzymes musculaires
(CPK, aldolase) et de lapriseen
1. Bases moléculaires,deux gènes sont
impliqués:a)Legène HJV (hémo-
juvénile) codé par le chromosome 1
responsable,lorsqu’il est muté,de la
forme laplus fréquented’hémo-
chromatosejuvénile (hémochroma-
tosede type 2A)[10].Une mutation
commune,G320V, aété retrouvée
parmi les 12 familles ayant fait
l’objet de ladescription originale,
mais de nombreuses mutations «pri-
vées»,c’est-à-diredes mutations
retrouvées dans une seule famille ou
chez des sujets isolés,ont été rap-
portées;b)Legène HAMP (Hepcidin
Anti-MicrobialPeptide). Localisé sur
le chromosome 19,cegène de l’hep-
cidine (hormone essentielle de ré-
gulation du métabolisme du fer [11])
comporte une mutation commune
G71Det plusieurs mutations privées,
responsables de l’hémochromatose
de type 2B[12].
2.Transmission :elle est de type
autosomal récessif.
3.Expression clinique:la surcharge
en fer s’exprime habituellement vers
l’âge de 20 ans,dominée par une
atteintecardiaqueet hypophysaire
(hypogonadisme hypogonadotrope).
Atteintehépatiqueet diabètepeu-
vent aussi survenir mais le pronostic
est avant tout fonction de l’atteinte
cardiaque. La forme liée à une
mutation de l’hepcidine serait par-
ticulièrement sévère.
4. Traitement:il est basé sur une
déplétion sanguine (saignées)in-
tensive,parfois associée,en cas
d’atteintecardiaque,àl’adminis-
tration parentérale de desferrioxa-
mine.
Chez le sujet de plus de 30 ans:deux
entités,exceptionnelles,peuvent être
en cause:
-Une surcharge en fer par mutation
en récepteur de la transferrine
de type 2 (ou hémochromatosede
type 3)[13]
1. Bases moléculaires:le récepteur de
la transferrine de type 2 est une pro-
téine homologuedu classique
récepteur de la transferrine de type1
mais àl’expression essentiellement
hépatocytaire. Ilest codé par le chro-
mosome 7.Plusieurs mutations ont
étédécrites.
137
••••••••

quidoit êtreévoquée en premier de-
vant une hyperferritinémie à satu-
ration normale de la transferrine.
L’existenced’un syndrome polymé-
taboliqueet le caractèrehabituelle-
ment modérédel’excès hépatique
en fer permettent de ladifférencier
d’une surcharge en fer par mutation
de laferroportine. Ilfaut toutefois
seméfier de l’impression d’atteinte
familiale quedonne le repérage
d’une hyperferritinémie chez des
membres de lafamille d’un sujet
présentant une hyperferritinémie
dysmétaboliquecar,en fait,il peut
seulement traduirelemême terrain
polymétaboliqueet non pas,comme
dans le syndrome ferroportine,la
transmission de lamutation selon
unmode dominant.
4. Traitement:il repose sur les sous-
tractions sanguines,dont la tolé-
ranceest inconstamment moins
bonne quedans les authentiques
hémochromatoses.
–Surcharge en fer par acéruloplas-
minémie héréditaire[19, 20]
1. Bases moléculaires:lacéruloplas-
mine est une protéine quiintervient
dans la sortie du fer des cellules
parenchymateuses par l’intermé-
diairede son activitéferroxidasequi
permet la transformation du fer fer-
reux (forme traversant les mem-
branes)enfer ferrique(forme per-
mettant lacaptation du fer sur la
transferrine plasmatique). Encas de
mutation du gène correspondant,
situé sur le chromosome 3, une
surcharge en fer viscérale diffuse se
développe aveclaparticularitéd’une
localisation cérébrale.
2.Transmission :elle est de type
autosomal récessif.
3.Expression clinique:elle se
démarquedes autres syndromes de
surcharge génétiqueenfer par
l’existenced’une anémie et par la
survenuede signes neurologiques
(signes extrapyramidaux,ataxie
cérébelleuse,dégénérescence réti-
nienne,démenceprogressive).
4. Traitement:les saignées sont en
règle contre-indiquées par l’anémie.
La desferrioxamine,en infusion
sous-cutanée prolongée,peut être
efficace.
SITUATIONS SANS SURCHARGE
VISCÉRALE EN FER
a)La maladie de Gaucher:au cours de
cette thésaurismosemacrophagique,
l’hyperferritinémie,de l’ordrede1000-
2000 µg/l,est un signe fréquent.Il
faut y penser notamment lorsquecette
hyperferritinémie, sans hypersidérémie
ni élévation de la saturation de la
transferrine, s’accompagne d’une splé-
nomégalie [21].
b)Le syndrome hyperferritinémie-
cataracte[22]:le piège est iciconstitué
par le caractèrefamilialdel’hyperferri-
tinémie,avecparfois réalisation de phlé-
botomies «pour hémochromatose»dans
certains membres de lafamille. Enfait,
ces phlébotomies s’avèrent avoir étéfort
mal tolérées (avecdéveloppement d’une
anémie),fer et saturation sont normaux,
et le diagnosticest le plus souvent
obtenu par un simple interrogatoire
orienté vers une histoireophtalmo-
logiquefamiliale «bruyante».Parfois,
cependant,l’histoirepersonnelle et/ou
familiale,peut n’êtrepas «parlante»,et
il convient alors de demander un
examen ophtalmologiquequipourra
découvrir une cataracteminime jusque-
làinsoupçonnée cliniquement.
c)Le syndrome d’activation macro-
phagique virale [23]:au cours
notamment d’une infection àEBV
ou àHSV, une hyperferritinémie
majeure(pouvant atteindreplusieurs
dizaines de milliers de µg/L)peut
s’observer.
d) Autres pathologies: une hyperferri-
tinémie très marquée (> 10 000 µg/l)
est une des marques biologiques de la
maladie de Still. Une hyperferritinémie
modérée,de mécanisme incertain,peut
êtreobservée au cours des hyperthy-
roïdies et des pathologies malignes,
viscérales ou hématologiques.
Au total,le pivot du diagnostic
d’hyperferritinémie est l’évaluation de
la saturation de la transferrine : sila
saturation est élevée,il s’agit (sous
réservequ’il n’y ait pas une cytolyse
majeured’origine hépatiqueou mus-
culaire) le plus souvent d’une hémo-
chromatoseliée au gène HFE (= de
type1) et,en cas de négativitédu test
HFE, d’une exceptionnelle hémochro-
matosenon liée àHFE (chez le sujet
jeune :hémochromatosede type 2,
2.Transmission :elle est de type
autosomal récessif.
3.Expression clinique: seules
quelques familles ont été rappor-
tées.Le tableau cliniqueest super-
posable àceluidel’hémochromatose
HFE, aveclapossibilitéd’une
expression plus précoce.
4. Traitement:il est basé sur les
saignées.
-Une forme particulièrede surcharge
en fer par mutation en ferroportine
(ou hémochromatosede type 4)
Eneffet, silamutation en ferroportine
donne classiquement lieu à une sur-
charge en fer avecnormalité(voire
abaissement)du taux de saturation de
la transferrine (voir ci-dessous),cer-
taines formes peuvent mimer phéno-
typiquement l’hémochromatoseavec
unprofil biologiquedes marqueurs
sériques martiaux indiscernables d’une
hémochromatosede type 1[14].
SURCHARGES GÉNÉTIQUES EN FER
AVEC NORMALITÉ
(OU QUASI-NORMALITÉ)DE
LA SATURATION DE LA TRANSFERRINE
Deux pathologies peuvent êtreen
cause:
–Surcharge en fer par mutation en
ferroportine [15-17]
1. Bases moléculaires:laferroportine
est une protéine impliquée dans la
sortie du fer des entérocytes ou des
macrophages.Son gène,appelé
SLC40A1,est portépar le chromo-
some 2.Sa mutation commune est
V162del. Plusieurs mutations pri-
vées ont étédécrites.Les mutations
en ferroportine semblent impliquées
dans la survenuedela surcharge en
fer des sujets noirs,qu’ils soient afri-
cains ou américains [18].
2.Transmission :elle est de type
autosomaldominant.
3.Expression clinique:il s’agit d’une
pathologie quiest relativement fré-
quente. Alors qu’une hyperferriti-
némie marquée peut survenir dès
l’enfance,l’expression clinique
semble de survenue tardiveet de
sévéritémodérée. La surcharge en
fer prédomine au niveau macro-
phagique. Son diagnosticdifféren-
tiel principalest l’hyperferritinémie
(ou hépatosidérose) dysmétabolique
138
••••••••

7.Mendler MH, Turlin B, Moirand R,
Jouanolle AM, Sapey T, Guyader D, Le
Gall JY,et al. Insulin resistance-asso-
ciated hepaticiron overload. Gastro-
enterology 1999; 117:1155-63.
8. Moirand R, Kerdavid F, LorealO,
Hubert N, Leroyer P, Brissot P, Lescoat
G.Regulation of ferritin expression by
alcohol in ahumanhepatoblastoma
cell line and in rat hepatocytecultures.
JHepatol 1995; 23:431-9.
9. Brissot P, Jouanolle AM, LeLanC,
LoréalO, Deugnier Y, David V.
Surcharges génétiques en fer non liées
àHFE.Gastroenterol Clin Biol 2005;
29:565-8.
10.Papanikolaou G, Samuels ME, Ludwig
EH, MacDonald ML, Franchini PL,
DubeMP,Andres L, et al. Mutations in
HFE2 causeiron overloadinchromo-
some 1q-linked juvenile hemochro-
matosis.Nat Genet 2004; 36: 77-82.
11.LorealO, Haziza-Pigeon C, Troadec
MB, DetivaudL, Turlin B, Courselaud
B, Ilyin G, et al. Hepcidin in iron
metabolism. Curr Protein Pept Sci
2005; 6: 279-91.
12.RoettoA, Papanikolaou G, Politou M,
AlbertiF, Girelli D, Christakis J,
Loukopoulos D, et al. Mutant antimi-
crobialpeptide hepcidin is associated
with severejuvenile hemochromatosis.
Nat Genet 2003; 33: 21-2.
13.CamaschellaC,RoettoA, Cali A, De
GobbiM, GarozzoG, CarellaM,
Majorano N, et al. The gene TFR2 is
mutated in anew type of haemochro-
matosis mapping to 7q22.Nat Genet
2000; 25:14-15.
14. DrakesmithH, Schimanski LM,
Ormerod E, Merryweather-Clarke AT,
Viprakasit V, Edwards JP,Sweetland
E, et al. Resistance tohepcidin is
conferred by hemochromatosis-
associated mutations of ferroportin.
Blood 2005; 106:1092-7.
chez le sujet plus âgé :hémochroma-
tosede type 3 voire rarement de type
4). Sila saturation est normale,l’hy-
perferritinémie peut traduire:a)dans
le cadredes pathologies fréquentes une
cytolyse, un syndrome inflammatoire
ou un syndrome polymétabolique; b)
dans le cadredepathologies rares sans
surcharge en fer, une maladie de
Gaucher ou un syndrome ferritine-
cataracte; c)dans le cadredes patho-
logies rares avec surcharge en fer, une
acéruloplasminémie héréditaireou une
mutation en ferroportine.
RÉFÉRENCES
1. Brissot P.Conduiteà tenir devant une
hyperferritinémie isolée. Gastroen-
térologie Pratique 2002; 138:1-3.
2.Aguilar Martinez P, Schved JF,Brissot
P.The evaluation of hyperferritinemia:
An updated strategy based on ad-
vances in detecting geneticabnorma-
lities.AmJGastroenterol 2005; 100:
1185-94
3.Pietrangelo A.Hereditary hemochro-
matosis—anew look at anold disease.
NEngl JMed 2004; 350: 2383-97.
4. HAS.Recommandations pour laPrise
en Charge de l’HémochromatoseHFE.
2005.
5. Guyader D, Jacquelinet C, Moirand R,
Turlin B, Mendler MH, Chaperon J,
David V, et al. Noninvasiveprediction
of fibrosis in C282Yhomozygous
hemochromatosis.Gastroenterology
1998; 115:929-36.
6.Brissot P, TroadecMB, LorealO.The
clinical relevanceofnew insights in
iron transport and metabolism. Curr
Hematol Rep 2004; 3:107-15.
15. MontosiG, DonovanA, TotaroA,
GarutiC, PignattiE, Cassanelli S,
Trenor CC, et al. Autosomal-dominant
hemochromatosis is associated witha
mutation in the ferroportin (SLC11A3)
gene. JClin Invest 2001; 108: 619-23.
16.Njajou OT,Vaessen N, JoosseM,
Berghuis B, vanDongen JW,Breuning
MH, Snijders PJ, et al. Amutation in
SLC11A3 is associated withautosomal
dominant hemochromatosis.Nat Genet
2001; 28: 213-14.
17.Pietrangelo A.The ferroportin disease.
Blood Cells Mol Dis 2004; 32:131-8.
18. GordeukVR, Caleffi A, Corradini E,
FerraraF,Jones RA, CastroO,
OnyekwereO, et al. Iron overloadin
Africans and African-Americans and
acommon mutation in the SCL40A1
(ferroportin 1) gene. Blood Cells Mol
Dis 2003; 31: 299-304.
19. Harris ZL, Klomp LW,Gitlin JD.
Aceruloplasminemia: aninherited neu-
rodegenerativedisease withimpair-
ment of iron homeostasis.AmJClin
Nutr 1998; 67:972S-977S.
20.LorealO, Turlin B, Pigeon C, Moisan
A, Ropert M, MoriceP, Gandon Y, et
al. Aceruloplasminemia: new clinical,
pathophysiologicaland therapeutic
insights.JHepatol 2002; 36:851-6.
21. Laine F, Guyader D, Turlin B, Moirand
R, Deugnier Y, Brissot P.[Hyper-ferri-
tinemiaand Gaucher disease].
Gastroenterol Clin Biol 1996; 20:512-
13.
22.FerranteM, Geubel A, Fevery J,
Marogy G, Horsmans Y, Nevens F.
Hereditary hyperferritinaemia-cata-
ract syndrome:achallenging diagnosis
for the hepatogastroenterologist.Eur
JGastroenterol Hepatol 2005; 17:
1247-53.
23.Fain O, Stirnemann J.Macrophage
activation syndrome. Rev Prat. 2004;
54:935-39.
139
••••••••
 6
7
8
6
7
8
1
/
8
100%