Les éliminations - Pages Persos Chez.com

L
LL
L
L
LL
L
L
LL
Le
ee
e
e
ee
e
e
ee
es
ss
s
s
ss
s
s
ss
s
é
éé
é
é
éé
é
é
éé
él
ll
l
l
ll
l
l
ll
li
ii
i
i
ii
i
i
ii
im
mm
m
m
mm
m
m
mm
mi
ii
i
i
ii
i
i
ii
in
nn
n
n
nn
n
n
nn
na
aa
a
a
aa
a
a
aa
at
tt
t
t
tt
t
t
tt
ti
ii
i
i
ii
i
i
ii
io
oo
o
o
oo
o
o
oo
on
nn
n
n
nn
n
n
nn
ns
ss
s
s
ss
s
s
ss
s
I
I_
_
L
Le
es
s
d
di
if
ff
fé
ér
re
en
nt
te
es
s
é
él
li
im
mi
in
na
at
ti
io
on
ns
s
A
A.
.
M
Mé
éc
ca
an
ni
is
sm
me
es
s
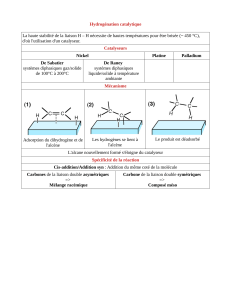
Une élimination consiste à former un alcène, en arrachant un proton et en
provoquant le départ d'un groupe partant. Différents mécanismes sont possibles :
q
q
Mécanisme concerté E
2
q
q
Avec un intermédiaire carbocationique E
1
q
q
Avec un intermédiaire carbanionique E
1CB
l
l
T
Th
hé
éo
or
ri
ie
e
d
de
e
l
l'
'é
ét
ta
at
t
d
de
e
t
tr
ra
an
ns
si
it
ti
io
on
n
v
va
ar
ri
ia
ab
bl
le
e
Il est possible d'imaginer que toutes les
éliminations sont en fait des E
2
, mais parfois à
tendance carbanionique (E
1CB
) lorsque le
groupe partant est mauvais mais le proton est
très acide, parfois à tendance carbocationique
(E
1
) lorsque le groupe partant est bon mais le
proton est peu acide.
B
B.
.
S
Sp
pé
éc
ci
if
fi
ic
ci
it
té
és
s
1
1)
)
P
Po
os
si
it
ti
io
on
n
d
de
e
l
l'
'é
él
li
im
mi
in
na
at
ti
io
on
n
L'élimination peut survenir comme précédemment sur deux carbones
adjacents ; on parle de 1,2-élimination. Elle peut se produit sur le même carbone,
pour former des carbènes ; on parle de 1,1-élimination.
H
X
Acide
Bon groupe
partant
B
H
X
Acide
Mauvais groupe
partant
B
X
C
ClCl
Cl H
OHC
Cl
Cl
Carbène
Très réactif
H
X
Peu acide
Bon groupe
partant
B
H
Force du groupe
partant
Acidité du
proton
Alcane
Alcène
E
2
E
1CB
E
1

Enfin elle peut survenir sur n'importe quelle carbone, à condition qu'ils soient
dans la bonne conformation. Ainsi il y en a quelques 1,3 et 1,4-éliminations.
2
2)
)
C
Co
om
mp
pé
ét
ti
it
ti
io
on
n
On observe souvent une compétition entre élimination et substitution
nucléophile. Cela dépend du rapport basicité/nucléophilie du nucléophile, mais aussi
des conditions de réaction, ainsi que des molécules mises en jeu.
3
3)
)
S
St
té
ér
ré
éo
os
sé
él
le
ec
ct
ti
iv
vi
it
té
é
L'élimination peut se produire en enlevant le proton et le groupe partant du
même côté, en syn, ou opposés, en anti. Cela conduit à deux énantiomères Z et E.
Le plus souvent on produit l'alcène E, plus stables.
4
4)
)
R
Ré
ég
gi
io
os
sé
él
le
ec
ct
ti
iv
vi
it
té
é
Selon la molécule, il est possible que l'élimination se fasse à plusieurs endroit,
il faut donc prendre en compte la régiosélectivité. Généralement, on forme l'alcène
le moins substitué, c'est la règle d'Haufmann.
C
C.
.
É
Él
li
im
mi
in
na
at
ti
io
on
ns
s
f
fa
av
vo
or
ri
is
sé
ée
es
s
On peut retenir que les E
1
et E
1CB
font intervenir des intermédiaires très
réactifs. Ainsi ces éliminations ne seront possibles que si les intermédiaires sont
stabilisés dans les conditions de la réaction.
1
1)
)
F
Fa
ac
ct
te
eu
ur
rs
s
f
fa
av
vo
or
ri
is
sa
an
nt
t
u
un
ne
e
E
E
1
1
Une E
1
est permise si la molécule est porteuse de groupements stabilisants la
charge positive du carbocation. Elle requiert un bon groupe partant, et se déroule
plus facilement dans des solvants polaires.
Elle n'est pas du tout influencée par la force de la base. Après formation du
carbocation, il y aura compétition entre E
1
et SN
1
.
2
2)
)
F
Fa
ac
ct
te
eu
ur
rs
s
f
fa
av
vo
or
ri
is
sa
an
nt
t
u
un
ne
e
E
E
1
1C
CB
B
Une E
1CB
est permise si la molécule est porteuse de groupements stabilisants
la charge négative du carbanion. Elle fonctionne très bien si un ammonium est
présent, un peu moins avec des groupements nitriles, cyanures et carbonyle, jamais
avec un halogène. Le proton arraché doit aussi être assez acide.
3
3)
)
F
Fa
ac
ct
te
eu
ur
rs
s
f
fa
av
vo
or
ri
is
sa
an
nt
t
u
un
ne
e
E
E
2
2
Une E
2
est influencée par la force de la base, du groupe partant et du solvant.
Par défaut, c'est-à-dire si les E
1
et E
1CB
sont impossibles, on fait préférentiellement
une E
2
(raisons énergétiques).
E
Énantiomère Z
cinétique
Énantiomère E
thermodynamique

4
4)
)
A
Au
ut
tr
re
es
s
f
fa
ac
ct
te
eu
ur
rs
s
l
l
B
Ba
as
si
ic
ci
it
té
é
d
du
u
n
nu
uc
cl
lé
éo
op
ph
hi
il
le
e
Pour une même molécule, on effectuera une réaction différente selon l'acidité
du nucléophile qui l'attaque.
Ex :
l
l
T
Ta
ai
il
ll
le
e
d
du
u
n
nu
uc
cl
lé
éo
op
ph
hi
il
le
e
Un nucléophile trop gros peut voir sa nucléophilie diminuer si l'encombrement
stérique est trop important.
Ex :
l
l
T
Te
em
mp
pé
ér
ra
at
tu
ur
re
e
On sait que la variation d'énergie libre varie avec la température d'un facteur
qui est la variation d'entropie. Dans le cas d'une élimination, on libère des
constituants de la molécule d'origine (H acide, groupe partant …) et on crée donc
plus d'entropie, contrairement à une substitution nucléophile. Ainsi plus la
température est élevée, plus l'élimination sera favorisée.
I
II
I_
_
G
Gé
éo
om
mé
ét
tr
ri
ie
e
d
de
es
s
p
pr
ro
od
du
ui
it
ts
s
f
fo
or
rm
mé
és
s
A
A.
.
O
Or
ri
ie
en
nt
ta
at
ti
io
on
n
d
de
es
s
é
él
li
im
mi
in
na
at
ti
io
on
ns
s
1
1)
)
E
E
1
1
Généralement on essaie de produire l'alcène le plus stable, qui est le plus
substitué.
l
l
P
Pr
ro
ob
bl
lè
èm
me
e
d
de
es
s
c
cy
yc
cl
le
es
s
Cl EtOH
Base faible
SN
1
OEt
Cl EtO Na
Base forte
E
2
H
+
Et
O
H
K OH
Br
Base très peu
encombrée
OH
K
Br
Base très
encombrée
SN
2
O
H
E
2

La géométrie d'un cycle (carbones tétraédriques) limite la formation d'un
alcène (carbones trigonaux).
l
l
L
Li
ib
br
re
e
r
ro
ot
ta
at
ti
io
on
n
e
et
t
e
en
nc
co
om
mb
br
re
em
me
en
nt
t
s
st
té
ér
ri
iq
qu
ue
e
Dans une E
1
, on forme presque toujours l'alcène le moins encombré, le plus
souvent de configuration E.
Ex :
2
2)
)
E
E
1
1C
CB
B
On enlève généralement le H le plus acide, et le moins encombré. On observe
que l'on fait souvent l'alcène le moins substitué avec une E
1CB
, c'est la règle de
Hofmann.
Ex :
3
3)
)
E
E
2
2
OH
E
1
H
+
Le moins substitué
défavorisé
Traces
Le plus substitué
favorisé
mais tensions de
cycle
Meilleure
hyperconjugaison
20 %
+
90 %
Le plus substitué
favorisé
O
H
Ph
H
+
E
1
Ph
H
CH
3
H
Intermédiaire le moins
encombré
Libre
rotation
Ph
+
Ph
E
majoritaire Z
minoritaire
N
H
HHH
Moins encombré et plus nombreux
cinétiquement favorable
E
1CB
NN
+
Alcène peu substitué

L'état de transition se rapproche déjà d'un alcène. On tend alors à former
l'alcène le plus substitué.
Pour la E
2
, les éliminations du groupe partant et du H acide peuvent se
produire du même côté de la molécule, on parle de syn-élimination, ou du côté
inverse, on parle d'anti-élimination.
En série acyclique, on effectue préférentiellement une anti-élimination, pour
des raisons stériques. Lorsque le groupe partant est très mauvais, on peut parfois
provoquer une syn-élimination.
En série cyclique, on fait toujours une anti-élimination, avec les substituants
en position axiale. En effet les orbitales doivent forcément être alignées pour que la
réaction se produise ( règle de Deslongchamps ).
B
B.
.
L
Le
es
s
s
sy
yn
n-
-é
él
li
im
mi
in
na
at
ti
io
on
ns
s
Ce type d'élimination ne peut se produire qu'en présence de bases
associées, dans des solvants peu polaires (ils doivent polariser les ions mais pas les
solvater).
Ex :-
HX
OMR
Interactions ioniques
augmentant la force du groupe
partant et l'acidité du H
+
R—OH
+
M—X
Organométallique
HX
ONaR
Interactions ioniques
augmentant la force du groupe
partant et l'acidité du H
+
R—OH
+
Na—X
Alcoolate
1
/
5
100%