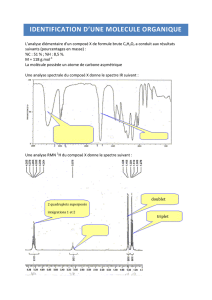
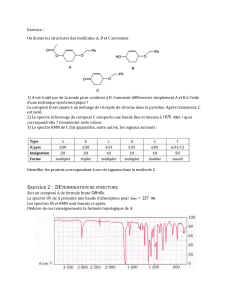
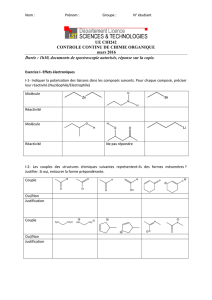
Document

CHAPITRE 13- ANALYSE SPECTROSCOPIQUE
1-Les Objectifs du chapitre.
2-Je maîtrise les définitions du chapitre
A-Spectroscopie UV-Visible et IR
•Définition de absorbance, transmittance, nombre d'onde.
•La loi de Beer-Lambert
•notions de déplacement chimique et constante de couplage.
Ce que je dois
connaître
•Calculer le nombre d'insaturations d'un composé organique.
•Savoir interpréter un spectre IR (Pics caractéristiques liés à une fonction chimique...)
•Identifier des noyaux d'hydrogène chimiquement (donc magnétiquement) équivalents
dans un composé.
•Exploiter les tables de données spectroscopiques pour dépouiller un spectre IR et RMN.
•Analyser un spectre de RMN 1H du premier ordre du type AmXp, AmMpXq
•Exploiter un spectre de RMN pour extraire les valeurs de déplacement chimique et les
valeurs des constantes de couplage.
Ce que je dois
savoir faire

a-La technique spectroscopique UV-Visible et IR
b-La spectroscopie infrarouge (IR) : Pourquoi les molécules absorbent-elles en IR ?
•Un rayonnement incident (intensité Io) de longueur d'onde (200nm<<800nm pour
spectre UV-visible) et (2 pour spectre IR) traverse un échantillon de
largeur une partie du rayonnement est absorbée, et l'intensité du rayonnement
transmis est I (avec I<Io). Un analyseur compare I et Io. Deux grandeurs permettent de
quantifier l'absorption du rayonnement incident: l'absorbance A et la transmittance
T.
Principe de
fonctionnement du
spectromètre UV-Visible
ou IR
•On définit l'absorbance par la relation:
; l'absorbance n'a pas d'unité.
•Loi de Beer-Lambert: en solution peu concentrée, l'absorbance A, est
proportionnelle à la concentration C de la substance absorbante, pour une longueur
d'onde et une température donnée: (avec appelé coefficient
d'absorption molaire de l'espèce absorbante en L.mol-1.cm-1, l est la largeur de la
cuve du spectrophotomètre (cm) et C (mol.L-1).
•Remarque: la loi de Beer-Lambert est additive pour une longueur d'onde donnée.
Absorbance: définition
Loi de Beer-Lambert
•On définit la transmittance d'une solution par la relation:
Transmittance

•La spectroscopie IR est une spectroscopie basée sur l'absorption des radiations IR par les molécules qui utilisent ce
type de rayonnement pour entrer en vibration
•il y a absorption en IR car les niveaux d'énergie vibrationnelle d'une molécule polyatomique sont quantifiés. La
molécule absorbe les rayonnements correspondant aux écarts énergétiques entre ces niveaux. Certaines transitions
entre ces niveaux sont possibles et conduisent alors à des modes propres de vibration de la molécule. . On distingue
deux types de modes propres de vibrations possibles pour une molécule polyatomique: les vibrations d'élongation et
les vibrations de déformation.
Remarque
•Vibration d'élongation: c'est une vibration affectant la longueur de la liaison entre deux atomes de la molécule.
•Vibration de déformation angulaire: c'est une vibration associée à une variation de l'angle entre deux liaisons de
valence.
•Remarque 1: les fréquences propres de vibration d'élongation sont en général plus élevées que celles de vibration de
déformation.
•Remarque 2:les fréquences de vibration d'élongation varie peu d'une molécule à l'autre. Cela permet d'identifier les
groupes fonctionnels dans les molécules organiques.
•Remarque 3: Seuls les modes propres de vibration conduisant à une variation du moment dipolaire de la molécule
sont actifs en IR.
Modes propres de
vibration d'une
molécule polyatomique
•Sur le spectre IR d'une molécule, on représente la transmittance en fonction du nombre d'onde de l'onde incidente.
•Sur le spectre IR, on observe des bandes plus ou moins larges car les transitions entre niveaux vibrationnels
s'accompagnent de transitions entre niveaux rotationnels. Ainsi, on n'observe pas de raies mais des bandes.
•on distingue deux régions dans le spectre IR:
•entre 4000 et 1300cm-1 = région d'observation des bandes (ou pics) de vibration d'élongation des groupes
fonctionnels de la molécule.
•entre 1300 et 600cm-1: région appelée empreinte digitale de la molécule ou l'on trouve les bandes de vibration de
déformation angulaires et quelques bandes d'élongation
Présentation du spectre
IR

B-La spectroscopie de RMN 1H (RMN du proton).
a-La technique spectroscopique de RMN
•La RMN 1H est une spectroscopie basée sur l'absorption d'un rayonnement
radio-fréquence par les noyaux des atomes d'hydrogène d'une molécule
placée dans un champ magnétique extérieur intense dans le spectromètre.
•un dispositif de détection et d'analyse permet d'analyser un phénomène
magnétique de relaxation se produisant après l'absorption. le spectre de
RMN de la molécule est obtenu après transformée de Fourier du signal.
Principe de
fonctionnement du
spectromètre de
RMN

b-La spectroscopie de RMN 1H : Pourquoi les noyaux des atomes d’hydrogène absorbent ils des rayonnements radio
•Les noyaux H possèdent un spin nucléaire l=1/2, ce qui leurs confère deux orientations possibles ( en présence d'un champ magnétique extérieur), caractérisées par un
nombre quantique de spin nucléaire ml= +1/2 ou -1/2. ces deux orientations donnent naissance à deux niveaux d'énergie magnétique différentes accessibles aux noyau H:
énergie liée à une orientation parallèle et celle liée à une orientation anti-parallèle du spin nucléaire par rapport au champ magnétique extérieur.
•En l'absence de champ magnétique, les deux niveaux d'énergie magnétique sont dégénérés. en présence d'un champ magnétique extérieur , il y a une levée de
dégénérescence des niveaux d'énergie magnétique du noyau: c'est l'effet Zeeman. Une transition devient possible entre ces deux niveaux si le noyau H reçoit un
rayonnement incident d'énergie égale à l'écart énergétique entre ces deux niveaux: il y a alors absorption du photon. ce type de transition énergétique fait intervenir des
radiations de fréquence allant de 40MHz à 600MHz (radio fréquences)
•Remarque: en présence d'un champ magnétique Bo, un proton ressent un champ magnétique B = Bo (1-, avec appelé constante d'écran (gandeur sans
dimension) dont la valeur dépend de l'environnement chimique du noyau. plus la densité électronique autour du noyau H est importante, plus la constante d'écran est
importante, le noyau est dit blindé. dans le cas contraire, le noyau H est dit déblindé (la constante d'écran est dans ce cas faible). La fréquence de résonance du noyau H
(fréquence du rayonnement incident qui est absorbé par le noyau) est donc fonction de son environnement chimique .
Niveaux d'énergie
magnétique des
noyaux
d'hydrogène
•La fréquence de résonance d'un noyau H dépend de la fréquence de travail de l'appareil de RMN utilisé. Afin de s'affranchir de cette dépendance, on définit le
déplacement chimique d'un noyau d'hydrogène par:
(en ppm, parties par million)
•dans cette relation, est la fréquence de résonance du noyau H étudié; est la fréquence de résonance des noyaux d'hydrogène d'un composé de référence (le
tétraméthylsilane, TMS (Si(CH3)4); est la fréquence de travail de l'appreil.
varie de 0 à 14ppm environ.
•plusieurs protons ayant le même environnement chimique, ont la même valeur du déplacement chimique. ils sont dits chimiquement équivalents, magnétiquement
équivalents ou isochrones.
Déplacement
chimique d'un
noyau H
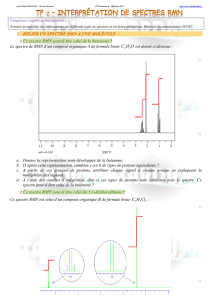
•Signal : sur le spectre de RMN d'une molécule, on représente l'intensité de l'absorption en fonction du déplacement chimique (qui est fonction de la fréquence de l'onde
incidente). chaque groupe de noyaux H isochrones est asssocié à un signal correspondant à une valeur de déplacement chimique déterminé.
•Courbe d'intégration: sur un spectre de RMN, la surface d'un signal est proportionnelle au nombre de noyaux qui résonnent pour donner ce signal. Une courbe
d'intégration est tracée sur le spectre, elle indique la proportionnalité entre la surface du signal et le nombre de protons à isochrones à l'origine du signal. La hauteur de
chaque palier de la courbe d'intégration est proportionnelleau nombre de noyaux associés.
•Constante de couplage: dans la plupart des molécules, les protons isochrones résonnent en donnant un groupe de pics appelé multiplet au lieu d'un pic (singulet). c'est la
conséquence d'une interaction entre le spin des protons isochrones et le spin des protons voisins: on parle de couplage spin-spin.
•Le couplage entre deux protons HAet HXséparés par n liaisons est caractérisé par la constante de couplage nJAX exprimée en Hz (variant de 0 à 20Hz).
•On observe essentiellent des couplages entres protons portés par des carbones voisins (ou vicinaux): on parle de couplage 3J . On peut observer des couplage 2J (protons
non isochrones portés par le même carbone (protons géminés) et des couplages 4J en général négligeables sauf pour les alcènes ou alcynes.
•Remarque: en général un H lié à un hétéroatome n'est pas couplé avec les autres H de la molécule.
Présentation d'un
spectre de RMN 1H
•Définition: le couplage est du premier ordre lorsque l'écart entre deux signaux associés à des noyaux 1H non équivalents est environ 10 fois supérieur à la
valeur de la constante de couplage J entre ces noyaux. le spectre est dit du premier ordre si
•Spectre de type AmXp: On a un spectre de type AmXp lorsque m noyaux isochrones A sont couplés avec p noyaux isochrones voisins X. La multiplicité du signal associé
aux m noyaux A est égale à (p+1) pics, et les intensités relatives des pics sont données par la règle du triangle de Pascal.
•Spectre de type AmMpXq: On a un spectre de type AmMpXq lorsque m noyaux isochrones sont couplés avec p noyaux isochrones d'une part, et q noyaux isochrones
d'autre part (non équivalents chimiquement aux précédents). l'l'allure des différents signaux s'interprete en étudiant successivement l'effet des différents couplages,
de la constante de couplage J la plus élevée à la moins élevée. pour les m noyaux H isochrones A le signal est un (p+1)-uplets de (q+1)-uplets si JAM different de JAX . Si
JAM environ égale à JAX alors le système se ramène à un spectre de type AmX(m+q) et on applique directement la règle des (n+1)-uplets.
Analyse d'un spectre
du premier ordre
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%