universite de nantes syndrome phaces : etude de sept cas et

0
UNIVERSITE DE NANTES
FACULTE DE MEDECINE
Année 2014 N° 021
THESE
pour le
DIPLOME D’ETAT DE DOCTEUR EN MEDECINE
DES de Pédiatrie
par
Jennifer HEBERT
née le 07/11/1985
Présentée et soutenue publiquement le 22/04/2014
SYNDROME PHACES : ETUDE DE SEPT CAS ET DISCUSSION
DIAGNOSTIQUE
Président : Monsieur le Professeur STALDER
Directeur de thèse : Madame le Docteur Hélène AUBERT

1
Remerciements
A Monsieur le Professeur STALDER, vous me faites l’honneur de présider ce jury et de juger
ce travail. Recevez ma sincère reconnaissance. Je vous remercie de m’avoir accueillie aux
consultations de dermatologie pédiatrique.
A Madame le Docteur Hélène AUBERT, pour avoir accepté de diriger ce travail. Je te
remercie pour ton aide et ton soutien précieux tout au long de ce travail. Merci de m’avoir fait
découvrir la dermatologie pédiatrique, ce fut un plaisir d’apprendre et de travailler à tes côtés.
A Madame le Professeur Gras-Le Guen, qui me fait l’honneur de juger ce travail. Je te
remercie pour tes enseignements et ton engagement tout au long de mon internat. Trouve ici
l’expression de mes sincères remerciements et de mon profond respect.
Au Professeur Rozé, qui me fait l’honneur de juger ce travail. Je vous remercie pour votre
disponibilité et vos conseils au cours de mon internat. Veuillez trouver l’expression de ma
sincère reconnaissance et de mon profond respect.
Aux pédiatres et aux médecins du CHU de Nantes et de la Maison des Adolescents de Cochin
avec qui j’ai eu l’honneur de travailler, je vous remercie sincèrement d’avoir contribué à ma
formation.
A mes co-internes, Merci pour les bons moments passés au cours de ces quatre années.
A mes parents, à Géraldine et Xavier, ainsi qu’à toute ma famille, pour votre soutien
inconditionnel.
A Pierre, merci pour ton soutien à toute épreuve.

2
Sommaire
I- Introduction p.3
I-A. Les hémangiomes p.5
I-A. 1 Epidémiologie p.5
I-A. 2 Clinique p.5
I-A. 3 Physiopathologie p.6
I-A. 4 Complications p.7
I-A. 5 Traitement p.8
I-B. L’hypothyroïdie congenital p.9
I-B. 1 Epidémiologie p.9
I-B. 2 Etiologies p.9
I-B. 3 Physiopathologie p.10
I-B. 4 Diagnostic p.12
I-B. 5 Traitement p.13
II- Présentation des cas cliniques p.14
II-A. Cas 1 p.14
II-B. Cas 2 p.17
II-C. Cas 3 p.18
II-D. Cas 4 p.20
II-E. Cas 5 p.23
II-F. Cas 6 p.25
II-G. Cas 7 p.27
Tableau 1 : Caractéristiques cliniques des 7 cas de notre série p.29
III- Discussion p.30
III-A. Le syndrome PHACES : revue de la littérature et comparaison p.31
de notre série
III-A. 1 Epidémiologie p.31
III-A. 2 Signes cliniques p.31
III-A. 3 Physiopathologie p.40
III-A. 4 Devenir, complications p.42
III-A. 5 Traitement p.44
III-B. Hémangiome et hypothyroïdie : syndrome PHACES p.45
III-C. Recommandations pour la pratique clinique p.51
IV- Conclusion p.52
Bibliographie p.53
Annexe 1 : Critères diagnostiques du syndrome PHACES p.57
Liste des abréviations p.58

3
I- INTRODUCTION :
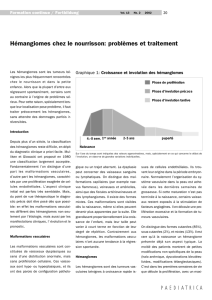
Les hémangiomes infantiles sont des tumeurs vasculaires très fréquentes qui touchent 4 à
10% des enfants. La grande majorité des hémangiomes sont isolés et de petite taille mais
certains sont associés à des syndromes poly-malformatifs, en particulier ceux de grande taille
ou de topographie particulière. Ainsi, les hémangiome segmentaires de la face sont associés
dans 20 à 31% des cas au syndrome PHACES [1, 2].
Le syndrome PHACE a été décrit en 1996 par Frieden and al [3]. Il associe un
hémangiome segmentaire de la face à une ou plusieurs des malformations suivantes :
anomalies de la fosse postérieure, anomalies vasculaires cérébrales intra et extra-crâniennes,
malformation cardiaque ou coarctation de l’aorte et anomalies ophtalmologiques. En 2002, les
anomalies de la ligne médiane et du sternum sont ajoutées aux atteintes extra-cutanées
possibles, formant ainsi l’acronyme PHACES syndrome.
Du fait de sa description relativement récente, les connaissances sur le syndrome
PHACES ont beaucoup évoluées ces dernières années. Les critères diagnostiques ont été
redéfinis en 2009 (annexe 1) [4]. La littérature compte actuellement 400 cas décrits sous
forme de cas cliniques ou de petites séries.
Nous présentons une série de 7 cas cliniques d’enfants présentant des hémangiomes
segmentaires du visage associés à des anomalies extra-cutanées. Ces enfants ont été pris en
charge dans les services de pédiatrie et de dermatologie du CHU de Nantes entre 2008 et
2013. Cinq des enfants ont un diagnostic avéré de PHACES syndrome, les 2 autres ont un
diagnostic possible, selon les critères révisés en 2009, puisqu’ils associent un hémangiome
segmentaire de la face et une ectopie thyroïdienne. Cette association n’a jamais été décrite

4
dans la littérature sans autres anomalies associées, d’où l’intérêt des deux derniers cas
présentés.
Après quelques rappels sur les hémangiomes et l’hypothyroïdie congénitale, nous
présenterons les cas cliniques. Ce travail a été l’occasion d’une revue de la littérature sur le
syndrome PHACES et sur l’association hémangiome et hypothyroïdie congénitale qui sera
présentée et discutée dans ce travail.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
1
/
60
100%