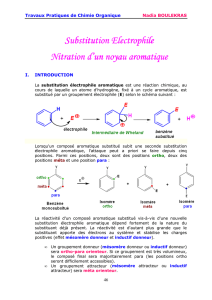
Substitution électrophile aromatique

-1-
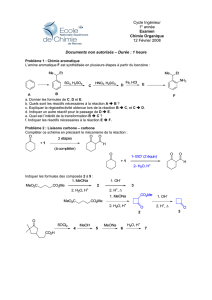
Chimie organique 7 :
Substitution électrophile aromatique
La famille des composés aromatiques, fréquemment rencontrés en chimie organique, compte parmi ses membres, le benzène
(C6H6), découvert en 1825 par Faraday.
A une époque où la chimie quantique et les méthodes spectroscopiques n’existaient pas, la détermination de sa structure a
suscité beaucoup d’intérêt et a conduit à de nombreuses propositions, toutes en cohérence avec la formule brute C6H6 :
3-Prismane Benzvalène Benzène Claus Benzène Dewar
Formules de Kékulé :
Proposées en 1865 à une époque où la délocalisation des électrons n’était pas connue
Validée par Pauling en 1928 (théorie de la mésomérie)
Les composés aromatiques sont caractérisés par une structure cyclique totalement conjuguée extrêmement stable
très difficile à modifier par oxydation ou par addition.
Seules les substitutions électrophiles aromatiques (SEAr) sont relativement faciles à réaliser car elles ne font pas
perdre le caractère aromatique à la molécule.
Dans cette réaction (SEAr), le cycle aromatique y joue le rôle de nucléophile et réagit avec un composé électrophile.
Ce chapitre s’intéresse trois aspects :
La notion d’aromaticité,
Les SEAr sur le benzène,
La SEAr sur les cylcles benzéniques déjà substitués.
1. Aromaticité
1.1. Exemples de composés aromatiques
- Dérivés du benzène : présence d’un noyau benzénique
O OH
OH
ON+
O
ONH2
OH

-2-
- Composés présentant plusieurs noyaux benzéniques accolés :
Naphtalène Phénanthrène Pyrène
- Cycles présentant des hétéroatomes
ON
H
N
Furane Pyrrole Pyridine
- Macrocycles à plusieurs dizaines d’atomes de carbone
H H
H
H
H
H
HH
H
H
H
H
HH
H
H
H
H
[18]annulène [24]annulène
Problématique : Quelle propriété ont en commun les composés aromatiques ? Qu’est-ce qui les caractérise ?
1.2. Stabilité des composés aromatiques
Les composés aromatiques sont extrêmement stables en raison de la délocalisation des électrons sur l’ensemble des atomes du
cycle. On constate qu’expérimentalement, il est très difficile de les faire réagir, en particulier lorsque la réaction envisagée
supprime la conjugaison au sein du cycle.
Pour évaluer la stabilité de la molécule, on détermine l’énergie de résonance, c’est-à-dire l’abaissement énergétique dû à la
délocalisation électronique en comparaison avec une molécule fictive les mêmes électrons mais sans délocalisation.
a) Evaluation de l’énergie de résonance
Méthode 1 : Enthalpies standard d’hydrogénation
On compare l’enthalpie standard de la réaction d’hydrogénation du benzène et on la compare avec celle du composé fictif
analogue au benzène mais sans délocalisation : le cyclohexa-1,3,5-triène.
1. Hydrogénation du cyclohexène :
Enthalpie standard d’hydrogénation d’une double liaison : ΔrH°1 = - 120 kJ.mol-1 (mesure expérimentale)
+H2=
2. Hydrogénation de l’hypothétique cyclohexa-1,3,5-triène obtenue en considérant qu’on hydrogène 3 doubles liaisons C=C
indépendantes : ΔrH°2 = 3 ΔrH°1 = - 360 kJ.mol-1 (valeur estimée théoriquement)
+3 H2=

-3-
3. Hydrogénation du benzène : Enthalpie standard de réaction : ΔrH°3 = - 210 kJ.mol-1 (mesure expérimentale)
+3 H2=
4. Report des valeurs sur un axe d’énergie (dans les deux cas, le produit obtenu est le cyclohexane) :
(Benzène)
(Cyclohexatriène)
(Cyclohexane)
E
Energie de résonance : Eres =
La délocalisation des électrons au sein du cycle aromatique est très stabilisante !
Ebenzène << Ecyclohexa-1,3,5-triène
Toute réaction faisant perdre le caractère aromatique à une molécule est très défavorable.
Méthode 2 : Méthode de Hückel
Chaque atome de carbone intervient dans le système π par une orbitale atomique 2p et un électron : le système π du
benzène possède donc 6 électrons.
Pour assurer la délocalisation, le benzène adopte une structure plane. De cette façon, les orbitales 2p intervenant dans le
système π sont parallèles entre elles (condition nécessaire à la délocalisation).
Le diagramme des OM π est le suivant :

-4-
Energie de résonance :
b) Inertie chimique des composés aromatiques :
Contrairement à ce que les formules de Kékulé laissent penser (écriture de doubles liaisons C=C), les cycles aromatiques ne
réagissent pas du tout comme les alcènes.
Les réactions d’addition sur les alcènes détruiraient les doubles liaisons et donc la conjugaison au sein du cycle… Elles sont donc
particulièrement défavorables et sont particulièrement difficiles à réaliser.
Les réactions d’addition :
Double liaison C=C
Cycle aromatique
Addition de Br2
Très facile
Impossible
Epoxydation
Facile
Impossible
Addition de H2
Très facile
Conditions douces
(T et P ambiantes)
Très difficile
Conditions très dures
(T et P très fortes)
Expl :
H2, [Ni]
1 bar, 25 °C
H2, [Ni]
100 bar, 200 °C
OH
O
KMnO4 cc
Chauffage
Seules les réactions de substitution sont réalisables facilement car elles
n’entraînent pas la disparition du système conjugué cyclique.

-5-
1.3. Propriétés spectroscopiques
- Spectroscopie RMN 1H : H aromatiques très fortement déblindés (δ ~ 7-8 ppm)
- Phénomène de courant de cycle :
Le champ magnétique imposé par l’appareil crée un courant d’électrons au sein du cycle. Ce courant d’électrons est
responsable d’un champ magnétique induit
qui s’oppose au champ extérieur
à l’intérieur du cycle.
Les atomes situés à l’intérieur du cycle sont soumis à un champ moins important
–
.
Les atomes situés à l’extérieur du cycle sont au contraire soumis à un champ plus important
+
: le champ
à
appliquer pour atteindre leur valeur normale de résonance
est donc moindre. Le déplacement chimique δ
correspondant est par conséquent plus grand.
1.4. Critère d’aromaticité de Hückel
Critère de Hückel : Un composé est aromatique s’il présente :
Sur un cycle plan ou des cycles plans accolés
4p + 2 électrons π (p : entier naturel)
Délocalisés sur l’ensemble des atomes du cycle
Composé
Conclusion
N
H
N
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
6
7
8
9
10
11
12
13
14
15
16
17
18
19
1
/
19
100%