Molécules et matériaux organiques Partie 3. Conversion par oxydo

-1-
Molécules et matériaux organiques
Partie 3. Conversion par oxydo-réduction
De l’alcène à l’époxyde… De l’époxyde au diol… De l’ester à l’alcool
Objectifs du chapitre
→ Notions à connaître :
Époxydation directe par un peroxyacide ; réactivité comparée des alcènes.
Ouverture des époxydes en milieu basique : mécanisme, élaboration de diols anti.
De l’acide ou de l’ester à l’aldéhyde ou à l’alcool primaire ; mécanisme schématique de la réduction
des esters.
→ Capacités exigibles :
Justifier l’usage d’une base comme l’hydrogénocarbonate de sodium dans l’élaboration de
l’époxyde.
Discuter de la régiosélectivité de l’époxydation sur un polyène.
Justifier la régiosélectivité et la stéréosélectivité de l’ouverture nucléophile d’un époxyde, en
l’absence d’activation par un acide de Lewis ou de Bronsted.
Interpréter la réduction d’un ester en alcool primaire en assimilant le réactif à un ion hydrure
nucléophile.
Identifier le produit de réduction d’un ester par un hydrure complexe, à l’aide de données fournies
(chimiques et/ou spectroscopiques).
Reconnaître ou proposer dans une stratégie de synthèse la conversion entre un ester et un aldéhyde
ou un alcool primaire.
Dans la continuité du programme de PCSI, le programme aborde la notion de conversion par oxydo-réduction en chimie
organique, à partir des deux groupes fonctionnels déjà rencontrés cette année :
Dérivé éthyléniques (double liaison C=C)
Acides carboxyliques et esters.
1. Quelques rappels sur l’oxydo-réduction
Nombre d’oxydation d’un élément au sein d’une structure :
Le nombre d’oxydation est la charge formelle fictive qui apparaît sur un atome après avoir procédé à
une répartition arbitraire de chaque doublet liant de l’édifice à l’atome le plus électronégatif de la
liaison.
Une oxydation étant une perte d’électron, le nombre d’oxydation indique bien le nombre d’électrons
qu’il a fallu ôter algébriquement à l’élément à partir de l’état fondamental (NO = 0).
Expl : Si NO(X) = +II, l’élément X est dans un état analogue à X2+ dans l’édifice : il a lui ôter fallu 2 e-.

-2-
Déterminer le nombre d’oxydation de chacun des atomes de carbone de la molécule d’acide éthanoïque.
Qu’est-ce qu’une oxydation ? Comment oxyder un atome de carbone d’une molécule organique ? Proposer un exemple
d’oxydation dans le programme de PCSI :
Qu’est-ce qu’une réduction ? Comment réduire un atome de carbone d’une molécule organique ? Proposer un exemple.

-3-
A partir de l’échelle ci-dessous, identifier la nature (oxydation ou réduction) que constitue la conversion :
D’un dérivé éthylénique en diol
D’un ester en alcool primaire
Associer une demi-équation électronique à chaque processus. Pourquoi parle-t-on de demi-équation ?
2. Accès aux époxydes
2.1. Présentation des groupes caractéristiques
Famille des époxydes = composés présentant un cycle oxacyclopropane (cyclopropane dont un atome
de carbone a été remplacé par un atome d’oxygène).
Oxacyclopropane :
Liaison péroxyde : Liaison simple O–O
Péroxyde d’hydrogène :
Famille des acides péroxycarboxyliques (plus simplement péracides) : Composé obtenu par oxydation
à l’eau oxygénée des acides carboxyliques.
Acide péroxyéthanoïque :

-4-
Le péracide le plus utilisé au laboratoire est le m-CPBA (acide méta-choroperbenzoïque).
Avantage : il est solide (donc facile à peser) et se conserve bien à basse température.
Les peracides sont nettement moins acides que les acides carboxyliques. Pourquoi ?
2.2. Action des péracides sur les dérivés éthyléniques (époxydation des alcènes)
Equation de la réaction :
(Conditions expérimentales usuelles : température ambiante dans le dichlorométhane.)
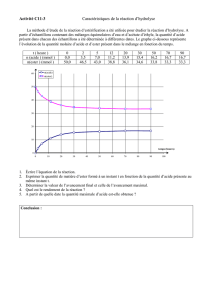
Réactivité comparée des alcènes :
krel 1 24 5,0.102 5,0.102 6,5.103 > 6,5.103
Interprétation :
Application : Régiosélectivité de l’action d’un péroxyacide

-5-
Aspect stéréochimique :
L’action d’un péracide sur une double liaison C=C est une syn-addition.
Prévoir les produits de l’action du mCPBA sur les deux stéréoisomères du but-2-ène :
L’action d’un péracide sur une double liaison dissymétrique est
2.3. Voies d’accès aux époxydes par des réactions intramoléculaires (PCSI)
Voie A : à partir des halohydrines.
Le terme « halohydrine » est synonyme d’α-halogénoalcools.
A partir des réactions au programme de PCSI, proposer une voie de conversion des halohydrines en époxydes.
Pourquoi est-il préférable d’utiliser une base relativement faible pour former l’époxyde ? Quelle autre réaction risquerait de
se produire en présence d’une base forte ?
 6
7
8
9
10
11
12
6
7
8
9
10
11
12
1
/
12
100%