Phases Préclinique et clinique

Pharmacologie Bo 04 C.L. BENHAMOU Semestre 4
1
Différentes phases de développement
d’un médicament
I. Phases Préclinique et clinique
Avant d’arriver à une molécule active, il faut découvrir une molécule, soit par découverte empirique,
soit par screening (sélection) de molécules, par des techniques de modélisation chimique.
Lorsqu’une molécule est découverte et qu’un brevet est déposé, le brevet protège pour 20 ans. On
dépose un brevet pour une molécule en décrivant son action. En effet, si l’on ne décrit pas un effet,
ce brevet ne le protège pas. Dans la vie d’une molécule, le développement dure 9 à 12 ans, et les
dossiers administratifs prennent 1 à 3 ans. Il reste donc en moyenne 8 ans de protection.
Exemple : Synthèse parallèle : 104 molécules
A débit intelligent : 100 molécules
Structure activée
Sélection biopharmacie pré
Modèles
10 atteignent le stade de test
Modélisation :
On veut bloquer un récepteur, se fixer dessus. On en connaît la conformation stéréologique.
On peut alors modéliser in silico (dans l’ordinateur), et on forme alors une série de molécules à
tester.
L’expérimentation se fait sur des cellules in vitro (exemple : cellules osseuses, myocardiques,
nerveuses…). Puis on retient les molécules qui n’ont pas d’effet secondaire.
La société a développé un logiciel : Selnergy. Ce logiciel permet de tester les molécules. Ils ont une
chimiothèque et une ciblothèque très développées. Ils essaient de faire concorder les deux selon une
base de données relationnelle plantes-molécules. Ils ont de plus une base de données
cristallographique. Cela permet de développer des molécules sur l’ordinateur, en entrant différentes
caractéristiques que l’on veut pour notre molécule.
Exemple de la viniférine : ils ont modifié l’e-viniférine pour en faire la z-viniférine, utilisable dans la
cosmétique. La z-viniférine devient spécifique d’un seul système biologique : la PDE-4.
A. Rapport bénéfice-risque
C’est un souci présent à toutes les étapes du développement. Il faut savoir si le procédé n’est pas
toxique. Il faut que le produit apporte quelque chose, sans avoir de propriété toxique, dès le début
du développement. Il faut aussi penser au rapport coût-bénéfice clinique. En effet, personne ne veut

Pharmacologie Bo 04 C.L. BENHAMOU Semestre 4
2
investir dans le développement d’un médicament qui ne pourra être utile qu’à peu de personnes. Le
rapport cout-bénéfice financier est aussi important, peu de personnes veulent investir à pertes.
Après l’expérimentation in vitro, il faut passer in vivo, sur de petits animaux (rat, souris, cobayes,
parfois lapins). Cependant, certains développements engagent de gros animaux avant de passer à
l’expérimentation humaine. Chez les petits animaux, on travaille sur quelques dizaines d’animaux,
chez les gros, on travaille sur un nombre plus limité : 2 à 5 individus. Souvent, on commence les
expérimentations sur de petits groupes avant de passer sur de plus gros.
On étudie d’abord la toxicité. Ensuite, on se penche sur la fonction de reproduction. Ensuite, on
regarde les propriétés mutagènes, les malformations… Ensuite, on regarde le potentiel cancérogène
de la molécule (leucémie, cancer…). Ensuite, on étudie la Pharmacodynamique (chemin du
médicament, sorties, répartition du produit, pharmacocinétique, pharmacodynamique…). Enfin, on
regarde les interactions médicamenteuses, il ne faut pas que le médicament empêche de prendre les
autres médicaments associés à la même pathologie, ou d’autres courantes.
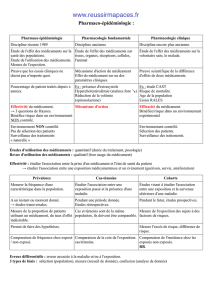
B. Les phases
Tout le développement animal est appelé Préclinique. On y étudie la toxicité à court terme, et la
toxicité à long terme. Elle dure en moyenne 3 ans. Ensuite, on en arrive à la phase clinique :
Phase 1 : volontaire sain, tolérance
Phase 2 : choix de dose
Phase 3 : étude pivot (efficacité, tolérance)
Phase 4 : études complémentaires.
1. Phase 1
Volontaire sain : jeunes, souvent étudiants en médecine, consentement éclairé, financement.
Première expérimentation humaine : précautions maximum en milieu hospitalier, peu de centres
(autorisation ministérielle du ministère de la santé, présence de gens qualifiés, d’équipes de
réanimation…), peu de sujets (environ 5 à 20). Ces études de phase 1 sont très encadrées. On y
évalue la tolérance, mais aussi la pharmacodynamique, et la pharmacocinétique. Dans les phases 1,
on donne parfois 1 seule dose, ou alors 2 doses et on fait un cross over (changement de doses en
croisement entre les groupes). On peut alors commencer le dose ranging, pour connaître les doses à
donner. Pour certains médicaments, la phase 1 est sautée, comme par exemple en cancérologie
(chimiothérapie).
2. Phase 2
La phase 2 est la phase du choix de la dose. L’administration se fait en versus placebo (sans produit
placebo = sans activité). Il faut des sujets malades, en grand nombre. La durée est variable. Elle a un
rôle capital pour définir la dose minimale efficace.
Placebo : produit inactif. Il est important de tester l’effet placebo. Tout médicament a un effet
placebo. Les gens, à partir du moment où ils avalent quelque chose, notent une amélioration. Il y a
aussi un effet nocebo, dès que les gens absorbent quelque chose, ils notent des effets indésirables.
3. Phase 3
Etudes pivots. Elles sont toujours développées contre placebo (est considéré comme non étique le
fait de soigner un malade avec un placebo alors qu’un médicament est démontré actif), et/ou contre

Pharmacologie Bo 04 C.L. BENHAMOU Semestre 4
3
produit de référence. Une seule dose, parfois 2. Nombre suffisant de malades (La méthode contre
produit de référence nécessite beaucoup plus de malade que la méthode placebo. Il faut préciser
l’évolution attendue, pour déterminer le risque β, et le nombre de malades). Echantillonnage clair
permettant de répondre à la question (type de malades, sexe, type de maladie, spécificité de
ciblage). Critères idem.
Généralement multicentriques : ± internationales, durée variable, études très encadrées.
Question principale : efficacité-tolérance
4. Phase 4
Questions complémentaires. Par exemple : sujet âgé, insuffisance rénale modérée,
enfant/adolescent, effets complémentaires du produit, impact épidémiologique.
Nombre variable, durée variable. Plus proche de la « vraie vie ».
Tout ça est différent des phases 4 à l’ancienne : ces phases 4 étaient plus des phases marketing,
commerciales.
II. Les Phases après la mise sur le marché
A. La mise sur le marché
1. L’autorisation de mise sur le marché
En France, l’agence qui s’appelait Agence du médicament, s’appelle aujourd’hui AFSSAPS. Elle
regroupe médicaments, mais aussi aliments et produits de santé en général. Il existe une agence
européenne, mais aussi une agence Américaine : la FDA (food and drug administration). En France, il
y a une soumission européenne, ou Française, mais il existe un inter soumission. Un médicament
accepté en France sera facilement accepté en Europe. Pour les autres pays, il faut passer par l’agence
nationale.
2. La durée globale
La mise sur le marché de la molécule dure 15 ans. Il faut ensuite 10 ans pour que la molécule passe
sur le marché du générique. Les couts déterminés par la firme de développement tiennent compte
de la durée de mise sur le marché.
3. Cout des différentes phases de développement
Phase 1 (quelques mois) : 1 à 4 millions d’euros pour 20 à 100 volontaires
Phase 2 (jusqu’à 2 ans) : 7 à 21 millions d’euros pour 100 à 1000 participants
Phase 3 (quelques années – 3 à 5 ans) : 50 à 100 millions d’euros pour 1000 à 5000 patients
Phase 4 (après l’AMM) : cout très variable non évaluable
4. Enregistrement
Autorisation de Mise sur le Marché : les experts de l’agence regardent tout le dossier, en
particulier la phase 3, et disent oui ou non, le médicament peut passer sur le marché

Pharmacologie Bo 04 C.L. BENHAMOU Semestre 4
4
Phase de Transparence : amélioration du service médical rendu. Juge de l’intérêt apporté au
marché et au service médical. Notion d’ASMR 1, 2, 3, 4. ASMR 1 : médicament qui
révolutionne le moyen de soigner une pathologie. ASMR 4 : médicament x qui soigne la
même pathologie sans rien changer.
Prix : négociation en fonction de l’ASMR. ASMR 1 : prix élevé. ASMR 4 : prix bas. L’agence
peut faire varier les prix en fonction du nombre de vente et de la durée de
commercialisation.
B. La pharmaco-vigilance
Elle recense les évènements indésirables une fois que le médicament est en circulation. Ils peuvent
apparaître même s’il n’est pas apparu en essai clinique. Elle a un rôle très important. Elle recense
surtout les accidents rares. Parfois, elle demande des études complémentaires.
C. Les génériques
Au bout de 10 ans, un médicament passe dans le domaine du générique, après l’AMM. Il prend un
nouveau nom dit « générique ». Ce nom vient en général du principe actif. Par exemple, le générique
pour le doliprane ou le dafalgan est le paracétamol. Pour se défendre contre les génériques, les
laboratoires peuvent proposer d’autres modes de présentation, des associations de produits,
fabriquer eux-mêmes les génériques, ce qui permet d’éviter les fabrications de génériques dans
l’immédiat. Cependant, cela nécessite un repassage de l’AMM.
D. Déremboursement ou moindre remboursement
Les médicaments sont classés en 3 groupes :
A : efficaces
B : moins efficaces
C : peu efficaces
Avec un pourcentage de remboursement en fonction du groupe. Par exemple : les veinotoniques.
E. Les essais
Les essais sont tous contrôlés. Ils sont différents des essais observationnels. En effet, on ne se
contente plus de noter les effets qui apparaissent. C’est différent des essais épidémio-
pharmacologiques, et des séries historiques. Ils sont caractérisés par le processus de randomisation
(tirage au sort). Ceci pour diminuer les biais, le plus tard possible à l’inclusion, de façon simple, en
bloc et en stratification.
Il existe des biais :
Simple aveugle : seul le malade ne sait pas ce qu’il reçoit.
Double aveugle : le malade et l’investigateur ne savent pas.
Triple aveugle : ni le malade, ni l’investigateur, ni l’interprétateur ne savent.
Il existe aussi des essais ouverts, qui ont moins de valeur, puisque tout le monde sait ce qu’il a.

Pharmacologie Bo 04 C.L. BENHAMOU Semestre 4
5
La Randomisation vient du vieux français Randon, qui est le mouvement que fait le cerf pendant qu’il
est chassé à cour. Ce mouvement aléatoire a été transformé en Random par les anglais, puis en
randomize, et randomization. Enfin, le mot est revenu en France sous le terme de randomisation.
Les effets mesurés dépendent des effets thérapeutiques, des effets du hasard, des effets des biais. La
randomisation a pour but de supprimer ou de diviser les biais. La randomisation se fait par tirage au
sort. On peut encore supprimer les biais par Stratification, après la randomisation. Par exemple, il y a
la stratification par centre, par sexe, par âge + sexe, par niveau de tension artérielle. En général, on
n’utilise pas plus de 2 strates. La levée d’aveugle se fait uniquement en cas de nécessité. Les codes de
randomisation sont possédés par les investigateurs, les coordinateur, les centres antipoison et les
promoteurs.
Les effets indésirables graves sont :
Le décès
Les évènements indésirables avec mise en jeu du pronostic vital
L’hospitalisation ou la prolongation de l’hospitalisation
L’invalidité ou l’incapacité
Tout évènement indésirable grave doit être déclaré dans les 24 heures au centre de vigilance et au
promoteur.
F. Pharmacovigilance après AMM
Elle se fait soit en études de phase 4, soit dans la pratique courante. Son rôle est la détection,
l’évaluation et la prévention des effets indésirables des médicaments. Avant l’AMM, on signale tous
les effets indésirables même si le rapport avec le médicament est douteux. On les classe en rapport
certain, probable, douteux ou improbable.
G. Le consentement éclairé
Il faut que le consentement éclairé soit écrit, le médecin doit lui expliquer ce qu’il va avoir comme
médicament, une fiche d’information doit être donnée longtemps à l’avance, le médecin doit laisser
un délai de réflexion au cobaye, et le cobaye peut retirer son consentement à tout instant, sans avoir
besoin de justification. Avoir des gens qui abandonnent l’essai, c’est quelque chose de très négatif, ça
veut dire qu’ils ne sont pas satisfaits du médicament.
H. CCPRB
Comité consultatif de protection des personnes se prêtant à la recherche biomédicale.
Anciennement, on les nommait comités d’éthique. Il y en a un par région. Ils veillent au respect du
secret médical et de la confidentialité. Il est généralement composé de 4 personnes de la recherche
biomédicale (dont 3 médecins), un médecin généraliste, 2 pharmaciens, un « éthicien » (religieux,
mère de famille), un sociologue, un juriste, un psychologue, un infirmier(e). Ils ont pour mission de
donner leurs avis sur l’information des personnes incluses ou à inclure, le consentement écrit, les
indemnités versées (que les salaires ne soient pas démesurés), la pertinence scientifique du projet,
l’adéquation entre les moyens et les buts du travail, l’existence d’une assurance, l’absence de
dangerosité inacceptable.
 6
7
8
9
10
11
6
7
8
9
10
11
1
/
11
100%