INFERENCE DE LA STRUCTURE GENETIQUE D`UNE POPULATION

INFERENCE DE LA
STRUCTURE
GENETIQUE D’UNE
POPULATION
TEST DU LOGICIEL STRUCTURE (VERSION
2.3.3) ET APPROCHE DES MODELES MIS EN
ŒUVRE PAR LE LOGICIEL
L A Y L A E L A S R I
E N S I M A G
2 E M E A N N E E – M M I S B I O I N F O
T R A V A U X D ’ E T U D E S E T D E R E C H E R C H E
T U T E U R S : O L I V I E R F R A N Ç O I S E T F L O R E N C E
M A R A N I N C H I

2
S O M M A I R E
1. La génétique des populations
2. Etudes théoriques de différents
modèles
3. Le logiciel MS
4. Le logiciel STRUCTURE
5. Expériences
6. Analyse des résultats et conclusion
7. Bibliographie

3
P R E A M BU L E
Notre travail a été effectué au laboratoire TIMC -Imag
sous la direction d’Oliver François et a concerné l’étude
d’une méthode de clustering implémentée par le logiciel
STRUCTURE.
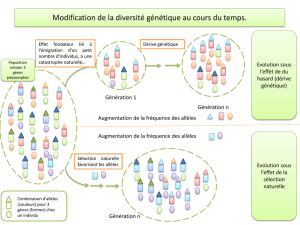
Lorsque l’on s’intéresse à l’évolutio n du patrimoine
génétique d’une espèce, la population est souvent prise
comme unité de base et il est utile de pouvoir répartir les
individus d’un échantillon au sein de populations.
Plusieurs approches sont alors envisag eables : on peut
chercher à grouper les individus d’un échantillon en
différentes populations ou chercher, à partir de la
connaissance d’un certain nombre de populations, la
provenance d’un individu d’origine inconnue.
Nous nous intéresserons ici à la première approche et à
la méthode dite de « clustering » ayant pour but de grouper
des individus en populations sur la base de leur génotype.
Un logiciel, largement utilisé en génétique des populations,
se propose d’implémenter cette méthode, le logiciel
STRUCTURE.
Notre travail consistera en l’étude de ce logiciel et des
algorithmes utilisés par celui-ci afin de déterminer la
portée et les limitations de ceux-ci. Pour cela, nous
procèderons de la sorte : nous utiliserons un logiciel
per mettant de générer des jeux de données génétiques à
partir de modèles prédéfinis, le logiciel MS. Nous ferons
traiter les données générées par ce logiciel par
STRUCTURE puis nous comparerons le résultat obtenu
avec celui attendu.
Nous commencerons par présenter les différentes
notions de génétique des populations qui seront utilisées
au cours de nos travaux futurs avant de présenter les
expériences réalisées avec les logiciels MS et STRUCTURE
avant de conclure quant à l’efficacité de la méthode de
« clustering » implémentée par le logiciel STRUCTURE.
Population :
ensemble
d'individus d'une
même espèce
vivante se
perpétuant dans
un territoire donné

4
L A G E N E T I Q U E D E S
P O P U L AT I O N S
OBJET DE LA GENETIQUE DES POPULATIONS
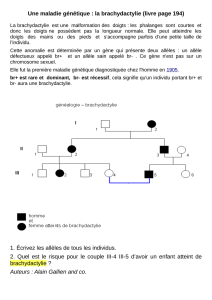
La génétique des populations traite de l’impact des différentes
forces évolutives (sélection, mutations, migration) sur la répartition de la
diversité génétique entre les populations et dans les populations.
La description de la composition génétique d’une population
diploïde se fait via différents indicateurs :
Les fréquences génotypiques : il s’agit des fréquences
des différents génotypes au locus considéré. L’ensemble
des fréquence génotypiques donne la structure
génotypique de la population pour ce locus.
Les fréquences alléliques (ou géniques) : il s’agit des
fréquences dans la population des différents allèles au
locus considéré. Leur connaissance donne la structure
allélique (génique) de la population.
L’ensemble formé par la structure génotypique et la structure
allélique d’une population constitue la structure génétique de la
population.
Cellule diploïde :
les chromosomes
qu’elle contient
sont présents par
paires.
Locus :
emplacement
physique précis et
invariable sur un
chromosome.
Génotype :
ensemble ou partie
donnée de la
composition
génétique d’un
individu.
Allèles d’un
gène :
les allèles d’un
gène
correspondent aux
différentes
versions de ce
gène (leurs
séquences de
nucléotides
diffèrent).

5
DESCRIPTION FORMELLE DE LA STRUCTURE
GENOTYPIQUE ET DE LA STRUCTURE ALLELIQUE D’UNE
POPULATION DIPLOIDE
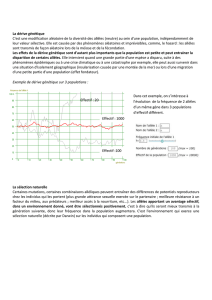
Considérons un gène A en un locus pour une population diploïde.
Structure génotypique en ce locus:
Où représente la fréquence des individus ayant le génotype AA,
est le nombre d’individus ayant ce génotype, et N le nombre total
d’individus.
Considérons désormais un gène A possédant de multiples allèles
A1
, …, An
en un
locus pour une population diploïde.
Structure allélique en ce locus :
Où représente la fréquence de l’allèle Ai en le locus considéré.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
1
/
22
100%