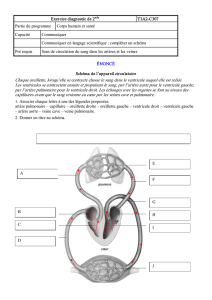
Cardiopathies Congénitales

Chapitre X, page 1
CARDIOPATHIES CONGENITALES
Les 10 points fondamentaux
1. La CIA est une cardiopathie congénitale fréquente longtemps bien tolérée
2. La CIA se complique de troubles du rythme auriculaire et d’une insuffisance cardiaque droite
3. La CIA réalise un shunt gauche droit comme la CIV. Dans la CIA l’importance du shunt est déterminée par la compliance des 2
ventricules, dans la CIV par les résistances à l’éjection des 2 ventricules.
4. La CIV est d’autant mieux tolérée que la communication est plus petite et que le souffle est important
5. Le traitement de la CIA et de la CIV reposent sur la fermeture de la communication
6. La tétralogie de Fallot associe une CIV large avec une sténose pulmonaire serrée ; c’est une cardiopathie cyanogène, dont la
sévérité clinique dépend du degré de sténose pulmonaire.
7. Dans la tétralogie de Fallot le pronostic actuel dépend du développement de la voie pulmonaire, difficile à reconstituer
chirurgicalement
8. Le canal artériel est maintenu ouvert pendant la grossesse par les kinines (les anti-inflammatoires risquent de le fermer, la
perfusion de prostacycline le maintient ouvert)
9. Le canal artériel réalise un shunt gauche droit dont la fermeture chirurgicale est simple.
10. La sténose pulmonaire se révèle d’autant plus tôt qu’elle est plus serrée : c’est au maximum une cardiopathie cyanogène de la
naissance, mais elle ne se manifeste parfois que pendant la vie adulte par des malaises d’effort (absence d’augmentation du débit
cardiaque).
Communication inter auriculaire (G Jondeau)
10% des cardiopathies congénitales chez les enfants,
40% des cardiopathies congénitales chez les adultes de plus de 40 ans
Anatomopathologie
3 types de communication inter-auriculaire peuvent être reconnues selon le siège de l'orifice:
Sinus venosus
haut situé, proche de l'abouchement des veines pulmonaires (souvent associé à un retour veineux pulmonaire anormal)
Ostium primum
bas , peut s'intégrer dans un canal atrio-ventriculaire plus complet (Anomalies des valves auriculo-ventriculaires et
communication inter-ventriculaire type admission)
Ostium secondum
foramen ovale (à différentier d'un foramen ovale persistant, présent dans 20% de la population adulte normale), souvent
associés à un prolapsus valvulaire mitral qui disparaît avec la cure de la communication inter-auriculaire
Lutembacher
rétrécissement mitral + communication inter-auriculaire
sinus veinosus
ostium secondum
ostium primum
VCI
VCS
VD
OD
tricuspide
Hémodynamique
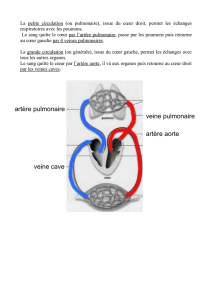
réalise un shunt des cavités gauches vers les cavités droites
L'importance de ce shunt dépend de 1) la taille de la communication inter-auriculaire qui transforme ± les 2 oreillettes en une cavité
unique, et 2) la compliance relative du ventricule droit et du ventricule gauche. En effet, c'est en diastole que survient le shunt, alors
que les valves mitrale et tricuspide sont ouvertes. Le ventricule qui est capable d'augmenter le plus son volume sans élever sa
pression (le plus compliant), est celui vers lequel va aller le + de sang. Le ventricule droit étant celui qui est le plus compliant est
celui qui se remplit le plus et le shunt est donc des cavités gauches vers les cavités droites.
A la naissance:
les résistances pulmonaires vont baisser progressivement et le ventricule droit va devenir moins épais (plus compliant). A l'inverse, la
résistance à l'éjection du ventricule gauche augmente et le ventricule gauche se muscle (et devient moins compliant). Le shunt des
cavités gauches vers les cavités droites augmente donc.

Chapitre X, page 2
tableau Clinique
Signes fonctionnels
Les enfants sont le plus souvent asymptomatiques, mais présentent parfois de nombreuses infection pulmonaire, reflet
du grand débit pulmonaire. C'est souvent chez les adultes que les signes fonctionnels apparaissent: troubles du rythme
auriculaire (reflet de la dilatation de l'oreillette droite), hypertension artérielle pulmonaire exceptionnelle (altérations
artériolaires pulmonaires, mais le complexe d'Eisenmenger est exceptionnel.) ou même une insuffisance cardiaque
(surcharge ventriculaire droite chronique, développement insuffisance mitrale). Enfin, existe un risque d'embolie
paradoxale (veine -> oreillette droite -> oreillette gauche -> systémique).
Signes physiques
Il existe un souffle systolique (de débit, réalisant un rétrécissement pulmonaire fonctionnel) et un dédoublement du B2
non modifié par la respiration (l'augmentation du retour veineux par l'inspiration est distribué dans les 2 oreillettes du
fait de la communication large)
Electrocardiogramme
L'hypertrophie ventriculaire droite (rSR' ou rsR' dans les précordiales droites) traduit la surcharge de volume ventriculaire droite.
Radiologiquement
Sont dilatées les cavités par où chemine le shunt (oreillette droite, ventricule droit, artère pulmonaire) et on retrouve une
hypervascularisation pulmonaire
Échographie
La visualisation directe de la communication inter-auriculaire par voie sous costale est possible (septum inter-auriculaire
perpendiculaire aux ultrasons). On recherche une anomalie associée, et étudie l'importance du shunt des cavités gauches
vers les cavités droites par l'étude du rapport des débits pulmonaire et aortique.
Cathétérisme
La sonde passe par la communication inter-auriculaire et permet de réaliser un cathétérisme gauche sans ponction artérielle. On
étudie le contenu en oxygéne veineux, artériel pulmonaire et artériel systémique pour calculer le rapport du débit pulmonaire sur
débit systémique (Qp/Qs).
On recherche des anomalies associées (notamment RVPA)
traitement
Correction chirurgicale entre 2 et 4 ans si Qp/Qs>1,5
suture simple ou patch de correction. La pose des ombrelles est en cours d'étude.
Le traitement médical comprend
La prévention Osler
Les antiarythmiques si apparaissent des tachycardies supra-ventriculaires qui peuvent compliquer la chirurgie comme
l'évolution spontanée.
Communications inter-ventriculaires (CIV) (G Jondeau)
Anatomopathologie (figure)
On distingue suivant la localisation de la communication inter-ventriculaire: 1) les CIV d'admission (par où le sang arrive, qui
s'intègrent souvent dans un canal atrio-ventriculaire), 2) les CIV musculaires qui sont souvent multiples, 3) les CIV d'éjection ou
infandibulaire, et 4) CIV peri-menbraneuses les + fréquentes
OD
12
3
4
Ao AP
Les CIV selon leur localisastion
1 CIV d'amission
2 CIV musculaire
3 CIV infandibulaire
4 CIV perimenbraneuse
Hémodynamique
L'importance de la fuite dépend
de la taille de la communication: si elle est large, tout se passe comme si existait une seule cavité ventriculaire, et la
pression dans les 2 ventricules est identique: le souffle est faible, mais le sang est éjecté vers le vaisseaux qui offre le
moins de résistance à l'éjection (en pratique l'artère pulmonaire, et le shunt est initialement des cavités gauches vers les

Chapitre X, page 3
cavités droites en l'absence de sténose pulmonaire). Par ailleurs, la pression pulmonaire est identique à la pression
aortique, et les artères pulmonaires vont se dilater et comprimer les bronches (infections pulmonaires, troubles de la
ventilation). Si la communication inter-ventriculaire est petite, les pressions ventriculaires droites systoliques restent
plus basses que les pressions ventriculaires gauches, et du sang va passer à grande vitesse (d'où souffle) du ventricule
gauche au ventricule droit.
Évolution
Les infections pulmonaires avec troubles de la ventilation sont fréquentes dans les communications inter-ventriculaires
larges, qui peuvent également se compliquer d'Eisenmenger, c'est à dire d'une hypertension artérielle pulmonaire,
conséquence de l'augmentation des résistances vasculaires pulmonaires (altération des artérioles par le haut débit et
l'hyperpression) avec inversion du shunt. Ceci est un phénomène irréversible, qui contre indique donc la fermeture de la
communication interventriculaire. Parfois, se développe une hypertrophie du ventricule droit produisant une obstruction.
Un tableau clinique proche de la tétralogie de Fallot apparaît ainsi dans 5 à 10% des cas. Enfin, en cas de
communication inter-ventriculaire située au contact des valves aortiques, peut se développer une insuffisance aortique
(5% des CIV).
Clinique
Petites CIV
maladie de Roger: gros souffle petit shunt (40%)
Le souffle réalise classiquement "beaucoup de bruit pour rien". Il est systolique (quand la différence de pression entre
ventricule gauche et ventricule droit est importante), mediothoracique, irradie en rayon de roue sur tout le thorax,
intense avec souvent un frémissement. Aucune autre anomalie n'est présente (notamment pas de retard de croissance).
L' évolution se fait spontanément vers la fermeture dans 50 à 75% des cas, parfois avec constitution transitoire d'un petit
anévrisme qui va couvrir la communication.
Les risques sont surtout 1) l'endocardite d'Osler, surtout après 18 mois, ce qui impose une prophylaxie antibiotique en
cas d'infection, et 2) la constitution d'une insuffisance aortique si la CIV est sous aortique.
Traitement: prévention Osler, surveillance bisannuelle
entre 3 et 6 ans, on évalue la pression artérielle pulmonaire et le rapport des shunts (Qp/Qs). Si la pression artérielle
pulmonaire est normale et le Qp/Qs inférieur à 1,5 on préfère suivre médicalement l'enfant du fait du risque opératoire
(notamment bloc auriculo-ventriculaire)
Grosses CIV
ventricule fonctionnellement unique, le shunt est fonction des résistances pulmonaire et systémique
A la naissance, la média des artères pulmonaires est épaisse, si bien que la résistance à l'éjection du ventricule droit est
élevée. Lors de la maturation des artères pulmonaires (premières semaines), le shunt va s'installer et entraîner 1) des
troubles de la croissance, 2) des effets cardiaques (souffle systolique si les pressions ventriculaires droite et gauche ne
sont pas parfaitement identiques, surcharge de débit de l'oreillette gauche, du ventricule gauche (avec roulement mitral
de débit), surcharge ventriculaire droite entraînant une hypertrophie qui peut conduire à une sténose pulmonaire
infundibulaire, insuffisance cardiaque, 3) des effets pulmonaires, témoins de la compression des bronches par les artères
pulmonaires distendues par la pression. Ces artères peuvent s'altérer et se fibroser pour aboutir à la constitution d'une
hypertension artérielle pulmonaire irréversible du fait de l'augmentation des résistances pulmonaires, inversant le shunt
(Eisenmenger)
Traitement
insuffisance cardiaque grave malgré traitement médical: la chirurgie s'impose
La fermeture de la CIV corrige complètement et définitivement la cardiopathie. Elle est donc préférée si le geste est
simple (communication inter-ventriculaire unique…). Sinon on réalise un cerclage de l'artère pulmonaire pour diminuer
l'hyperdébit et l'hyperpression pulmonaires puis on ferme la ou les CIV 1 an plus tard.
Si l'enfant est bien sous traitement médical
On attend 2 ans pour réaliser une cure complète si la CIV ne s'est pas spontanément fermée.
Cette attitude est à pondérer en fonction des données de chaque cas
Si existe une insuffisance aortique (CIV sous aortique), l'attitude est plus chirurgicale du fait du risque d'aggravation de
la fuite aortique. Si les CIV sont multiples, le geste chirurgical est plus complexe et souvent incomplet, et on tente de le
retarder autant que possible dans l'espoir qu'une partie des CIV se ferment et que le coeur soit plus grand (croissance).
La CIV peut s'associer à d'autres anomalies, qui vont modifier l'attitude. Enfin en cas de syndrome d'Eisenmenger, il ne
faut pas opérer et la seule alternative au traitement médical est la transplantation coeur-poumons.

Chapitre X, page 4
Tétralogie de Fallot (T4) (G Jondeau)
anatomie pathologique
10% des cardiopathies congénitales
Elle s'appelle tétralogie car elle réunit 4 éléments: 1) une communication inter-ventriculaire large réalisant l'égalité de pression entre
ventricule droit et ventricule gauche, qui résulte du malalignement du septum, 2) Le chevauchement de l'aorte qui résulte également
du malalignement du SIV, 3) une obstruction de la voie pulmonaire, qui est l'élément fondamental du pronostic et comprend souvent
a) une sténose infundibulaire, musculaire, donc susceptible de varier dans le temps. b) une sténose valvulaire avec au maximum
occlusion complète; l'arbre pulmonaire est alors vascularisé par le canal artériel. c) une sténose des branches des artères pulmonaires,
et 4) une hypertrophie ventriculaire droite du fait de la pression élevée qui règne dans cette cavité.
Peuvent s'y associer une anomalies des artères coronaires (important pour le chirurgien), et d'autres anomalies cardiaques.
Hémodynamique
Comme la communication inter-ventriculaire est large, l'importance relative des débits est liée au rapport des résistances à l'éjection
des ventricules: plus la sténose pulmonaire est serrée, moins le débit pulmonaire sera élevé, et donc plus le shunt des cavités droites
vers les cavités gauches sera important et la cyanose sévère. A l'inverse, si l'on augmente les résistances systémiques, la part qui va
vers les poumons va augmenter. Chez les enfants qui ont une sténose pulmonaire importante, surviennent des malaises, avec accès de
cyanose et désaturation très sévère, parfois avec acidose, surtout après les efforts, c'est à dire lorsque les vaisseaux périphériques sont
dilatés (diminution de la résistance à l'éjection du ventricule gauche). L'hypoxie et l'acidose peuvent aggraver la sténose pulmonaire
en entraînant une contraction de la sténose musculaire pulmonaire. Ces malaises peuvent être mortels, et les enfants les limitent en
augmentant les résistances systémiques par une position accroupie, avec les bras également pliés, ce qui tend à équilibrer la résistance
à l'éjection des 2 ventricules.
L'hypoxie entraîne la sécrétion d'érythropoiétine qui produit une polyglobulie, jusqu'à un niveau permettant une oxygénation
tissulaire suffisante. Si la polyglobulie est insuffisante pour corriger l'hypoxie chronique, la concentration de l'hémoglobine continue
d'augmenter jusqu'à entraîner hyperviscosité, avec ses risques de thrombose et de saignement… Parallèlement à la polyglobulie, se
développe un hippocratisme digital (déformation des doigts en baguette de tambour). Enfin, la polyglobulie résulte du passage du
sang veineux systèmique dans l'aorte, et l'on imagine aisément qu'un embol septique peut suivre le même chemin et entrainer un
abcès cérébral du fait de l'absence de "filtre" pulmonaire.
clinique
Ces enfants sont très "miséreux", du fait de l'hypotrophie et de la cyanose. Existe à l'auscultation un souffle systolique de sténose
pulmonaire, d'autant plus intense que la sténose pulmonaire est moins serrée (c'est à dire d'autant plus intense que plus de sang passe
à travers la sténose pulmonaire). Lors des accès d'hypoxie, liés à la diminution du débit pulmonaire, le souffle diminue ou peut même
disparaître. Le B2 est unique car seule la composante aortique est audible.
l'ECG retrouve une hypertrophie ventriculaire droite et auriculaire droite.
L'aspect radiographique est typique et réalise le coeur en sabot: l'arc pulmonaire manque et le ventricule droit saille particulièrement.
La vascularisation pulmonaire est pauvre.
L'echocardiographie permet le diagnostic de certitude.
Le cathétérisme permet de faire le bilan exact des lésions, et l'angiographie apprécié la voie pulmonaire.
Le traitement idéal est la correction complète de l'anomalie
On la réalise de plus en plus tôt, même si l'âge idéal est de 2 ans, elle est actuellement possible à partir de 6 mois. Si l'enfant ne peut
attendre cet âge ou présente une forme peu favorable à la chirurgie (essentiellement atrophie de la voie pulmonaire), on peut réaliser
une anastomose de Blalock, entre la sous clavière gauche et l'artère pulmonaire éventuellement avec un patch de Goretex.

Chapitre X, page 5
Persistance du canal artériel (G Jondeau)
Anatomie pathologique
10-15% des cardiopathies congénitales, plus fréquente chez les filles (2-3 filles / garçon). Le canal artériel réalise une communication
entre l'aorte après le départ de la sous clavière gauche et le tronc de l'artère pulmonaire (Permet, pendant la vie foetale, le passage du
sang de l'artère pulmonaire vers l'aorte descendante, c'est à dire diminue la vascularisation du poumon non encore fonctionnel). Il
réalise un shunt dont le sens et l'intensité dépendent de 1) la différence de pression entre l'aorte et l'artère pulmonaire, 2) la taille du
canal, et 3) la longueur du canal. Sa structure est musculaire et le canal est maintenu ouvert par la sécrétion de prostaglandines en
réponse à la faible PO2 pendant la vie foetale (risque Aspirine pendant grossesse). Lors de la naissance: l'oxygénation des poumons
entraîne une augmentation de la PO2 et ainsi une diminution de la synthèse de prostaglandines, si bien que le canal artériel se
vasoconstricte intensément (en altitude le canal se ferme plus lentement). Dans un 2° temps, il se fibrose et s'oblitère complètement
(quelques semaines). Ceci ne peut survenir que si le poumon est assez mature (le canal persiste chez les prématurés, et peut avoir un
rôle dans les insuffisances cardiaques et respiratoires du prématuré). Sa persistance peut être bénéfique lors de certaines cardiopathies
congénitales, car il peut permettre une vascularisation pulmonaire (atrésie pulmonaire), ou une vascularisation de l'aorte ascendante
(coarctation très serrée) ou de toute l'aorte (hypoplasie du VG): on le maintient ouvert alors après la naissance par perfusion de
prostaglandine E1.
Hémodynamique
Le flux sanguin à travers le canal dépend de la différence de pression entre l'aorte et l'artère pulmonaire. Au début de la vie les
résistances pulmonaires baissent, (+ lentement chez le prématuré), et le shunt s'installe progressivement après la naissance. Lorsque
le débit à travers le canal est élevé, il entraîne une augmentation du débit à travers oreillette et ventricule gauches (surcharge
volumétrique) et une diminution de la pression artérielle diastolique du fait du reflux de sang de l'aorte dans l'artère pulmonaire.
Clinique
Chez le prématuré
Les signes périphériques sont proches de ceux observés dans l'insuffisance aortique, avec élargissement de la
différentielle aux membres inférieurs. A l'auscultation on entend un souffle systolique ou systolo-diastolique sous
claviculaire gauche. Enfin on peut observer des signes de décompensation cardiaque ou/et pulmonaire.
Le traitement repose en règle sur l'indometacine (inhibiteur des prostaglandines), et en cas d'échec (10%) sur la ligature
chirurgicale du canal.
Chez les nouveau-nés à terme et les enfants
Les signes pulmonaires à type de pneumopathies à répétition ou de dyspnée d'effort sont parfois la circonstance de
découverte. A l'auscultation on retrouve un souffle continu à renforcement systolique sous claviculaire gauche, mimant
"une locomotive dans un tunnel", frémissant. Peuvent s'y associer des souffles de débit mitral et aortique. En périphérie,
on retrouve un élargissement de la différentielle (reflux de l'aorte dans l'AP).
A l'ECG: hypertrophie ventriculaire gauche diastolique puis systolique
Radiologiquement, on retrouve la dilatation de l'oreillette gauche et du ventricule gauche, ainsi que de l'aorte ascendante
et du tronc de l'artère pulmonaire, avec une hypervascularisation pulmonaire
L'echocardiographie permet le diagnostic et de quantifier le shunt.
Le traitement est chirurgical et simple (pas de circulation extra-corporelle). La pose d'ombrelles, endocavitaire, est en
cours d'étude. Le risque de l'évolution spontanée est l'endocardite et le développement d'une hypertension artérielle
pulmonaire (avec au maximum Eisenmenger) qui ne devraient plus se voir.
Rétrécissement pulmonaire (RP) (G Jondeau)
Anatomopathologie
7% des cardiopathies congénitales
La sténose pulmonaire peut être très modérée, intermédiaire ou très sévère avec subostruction pulmonaire. La simplicité du traitement
correcteur dépend du type de sténose. Les sténoses valvulaires se corrigent simplement, mais les anomalies de l'infandibulum
pulmonaire sont plus difficile à corriger (chirurgie).
RP très serré révélé en période néo natale
La sténose pulmonaire gène l'éjection du ventricule droit, dont le développement peut également être anormal. L'oreillette droite ne
peut donc se vider dans le ventricule droit et "force" le foramen ovale qui ne va pas se fermer. Il s'agit alors d'une cardiopathie
cyanogène. Si le canal artériel reste perméable, il va permettre une amélioration du flux pulmonaire.
Cliniquement
Il existe une cyanose dès la naissance, avec radiologiquement cardiomégalie (à la différence de la tétralogie de Fallot) et
hypoperfusion pulmonaire. A l'ECG, on observe une hypertrophie ventriculaire gauche et non une hypertrophie
ventriculaire droite (à la différence de la tétralogie de Fallot).
traitement
dilatation par voie endocavitaire, si le ventricule droit est assez développé
Chez l'enfant
Le plus souvent aucun signe fonctionnel, mais parfois une dyspnée d'effort modérée, une cyanose modérée, des syncopes d'effort (le
débit cardiaque ne peut augmenter au cours de l'effort). A l'auscultation, on retrouve un souffle systolique de sténose pulmonaire
(foyer pulmonaire), le plus souvent frémissant, avec claquement lors de l'ouverture de la valve pulmonaire (click d'éjection).
 6
7
8
6
7
8
1
/
8
100%