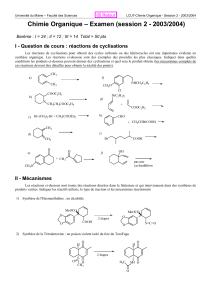
1/32 BiochAA. cours Métabolisme des acides aminés – LEMAIRE

BiochAA. cours
Métabolisme des acides aminés – LEMAIRE.
1/32
Biochimie – Métabolisme particulier
des acides aminés.
Métabolisme de la Sérine et de la glycine.
Acides aminés non-indispensables, glucoformateurs et interconvertibles.
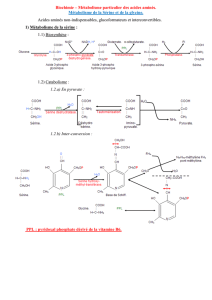
1) Métabolisme de la sérine :
1.1) Biosynthèse
1
:
1.2) Catabolisme :
1.2.a) En pyruvate :
1.2.b) Inter-conversion :
1
PPL : pyridoxal phosphate dérivé de la vitamine B6.
Glucose.
Glycolyse.
COOH
H–C–OH
CH2OP
Acide 3-phospho
glycérique.
NAD+
NADH, H+
Glutamate.
COOH
C=O
CH2OP
Acide 3-phospho
hydroxy-pyruvique..
3-phospho glycérate
déshydrogénase.
α-cétoglutarate.
Transaminase.
PPL
COOH
H–C–NH2
CH2OP
3-phospho-sérine
Pi
H2.
COOH
H–C–NH2
CH2OH
Sérine.
Phosphatase.
O
CH
Base de Schiff.
CH2O
Sérine hydroxy-
méthyl-transférase.
H2O
PPL
COOH
H–C–NH2
CH2OH
Sérine.
N
CH2OP
CH3.
HO
CH2OH
CH–COOH
N
CH
PPL
N
CH2OP
CH3.
HO
FH4.
H2O
N5-N10-méthylène FH4
pont méthylène.
CH2–COOH
N
CH
N
CH2OP
CH3.
HO
COOH
H–C–NH2
Glycine.
Sérine déshydratase.
H2O
COOH
C–NH2
CH2
Tautomérisation.
PPL
COOH
H–C–NH2
CH2OH
Sérine.
COOH
C=NH
CH3
Imino-
pyruvate.
NH3.
COOH
C=O
CH3
Pyruvate.
H2O
Déshydro
-sérine.

BiochAA. cours
Métabolisme des acides aminés – LEMAIRE.
2/32
1.2.c) Ethanolamine :
Lorsqu’il existe un défaut de fermeture du tube neural (22 à 29ème jours) lors de la vie
embryonnaire, on est en présence d’un spina bifida.
On peut dépister cette anomalie par échographie ou par chromatographie : on prélève du
liquide amniotique, si la fermeture est effectuée correctement, il n’y aura pas présence
d’acétylcholine, en revanche, en cas de défaut de fermeture, on retrouvera de l’acétylcholine dans le
liquide amniotique.
Le test de dépistage de la présence d’acétylcholine est qualitatif. On effectue trois tests, un
test témoin (LCR contenant à coup sur de l’acétylcholine), une bandelette avec le prélèvement
amniotique et une 3ème avec ce même prélèvement auquel on ajoute un inhibiteur de l’acétylcholine.
On ajoute le substrat de l’acétylcholine avec lequel elle va réagir, qui prouve donc sa présence
et on obtient :
1.3) Participation à la formation de produits spécialisés : par exemple des sphingolipides.
Ethanolamine.
CH2OH–CH2–NH2.
CAT.
(Choline Acétyl Transférase).
CO2.
PPL
COOH
H–C–NH2
CH2OH
Sérine.
CH3.
COOH
CH–NH2
CH2
CH2
S+––ADO
CH3
R–CH2OP
Acide phosphatidique.
R–CH2OPOCH2–CH2–NH2.
Phosphatidyl-éthanolamine.
S-adénosyl-
méthionine
= SAM.
R–CH2OPOCH2–CH2–NH–CH3.
Phosphatidyl-méthyl-éthanolamine.
CH3.
CH3
R–CH2OPOCH2–CH2–N–CH3.
Phosphatidyl-diméthyl-éthanolamine.
CH3
R–CH2OPOCH2–CH2–N+–CH3.
CH3
Phosphatidyl-choline.
CH3.
RCH2OP
CH3
HOCH2–CH2–N+–CH3.
CH3
Choline.
Acétyl-CoA.
CoA-SH.
O CH3
CH3–C–OCH2–CH2–N+–CH3.
CH3
Acétylcholine.
Acétylcholine
estérase.
Acétate.
+
Choline.
L.C.R.
L.C.R.
Prélèvement
amniotique
Prélèvement amniotique
+ inhibiteur.
Prélèvement amniotique
+ inhibiteur.
Prélèvement
amniotique.
Sujet atteint.
Sujet sain.

BiochAA. cours
Métabolisme des acides aminés – LEMAIRE.
3/32
2) Métabolisme de la glycine :
2.1) Biosynthèse :
2.1.a) A partir de la sérine : même schéma que tout à l’heure.
2.1.b) A partir de CO2 et NH3 :
Cette réaction est parfaitement réversible, mais
elle fonctionne surtout dans le sens du catabolisme.
2.1.c) A partir de l’acide glyoxylique :
2.2) Le catabolisme de la glycine :
1ère voie : Catabolisme de la glycine avec formation de pyruvate (acide aminé
glucoformateur).
2ème voie : catabolisme de la glycine CO2 + NH3 + NADH, H+ (cf. synthèse glycine).
Pathologie : hyperglycinémie sans cétose : accumulation dans le plasma de glycine. C’est une
maladie très grave qui provoque la mort de l’enfant très jeune. La cause est une défaillance de la
glycine synthase (qui ne détruit plus la glycine).
Hyperglycinémie avec cétose :???
2.3) Participation à la formation de produits spécialisés :
Elle participe aux réactions de détoxification (dans le foie) en se fixant à de grosses molécules très
hydrophobes, pour permettre leur élimination par la bile.
2.3.a) Formation de l’hippurate (découvert dans l’urine de cheval) ?
Dans les excès en glycine, on
essaye d’augmenter les apports en
benzoate pour former plus
d’hippurate élimination urinaire.
CO2 + NH3 + NADH + H+
N5N10méthylèneFH4
FH4
PPL
COOH
CH2–NH2.
+ NAD+
Glycine.
Glycine synthase
mitochondriale.
Xylulose 5P.
Acide glycolique
NH4+
.
HOCH2–COOH
Glycine.
NADH, H+
O
HC–COOH
Acide glyoxylique
COOH
CH2–NH2.
NAD+
FADH2.
FAD ou FMN
Transamination.
PPL
H2O.
NAD+
NADH, H+
O
HO–C–COOH
Acide oxalique.
Calculs.
COOH
ATP.
AMP + PPi.
CoA-SH.
CO~S-CoA.
Glycine.
CoA-SH.
Benzoate
.
Benzoyl-CoA.
COOH
CO–NH–CH2.
Acide
hippurique.

BiochAA. cours
Métabolisme des acides aminés – LEMAIRE.
4/32
2.3.b) γ-glutamyl-cystéine-glycine : glutathion.
2.3.c) Synthèse de la créatine :
Chaque jour une petite
partie de la créatine se cyclise en
créatinine. Cette quantité est
proportionnelle à la masse
musculaire. Une quantité de
créatinine trop élevée traduit un
taux de créatine anormalement
supérieur. Dopage ???.
2.3.d) Synthèse de l’hème (par l’ALA synthase
1
) :
L’ALA déshydratase cytoplasmique, est
constitué de zinc. La présence de plomb
l’inhibe et la réaction ne peut être catalysée. La
pathologie associée à cette intoxication au
plomb est appelée saturnisme.
1
ALA synthase : Acide δ-amino-lévulinique synthase.
Arginine.
COO–
H–C–NH3+
CH2
CH2
CH2
NH
HN=C–NH3+.
Arginine-glycine
transamidinase.
COOH
CH2–NH2.
Glycine.
COO–
H–C–NH3+
CH2
CH2
CH2
NH2
Ornthine.
NH2
H2N+=C NH–CH2–COOH.
Acide guanidoacétique.
= glycocyamine.
REIN.
SAM
SAH
Guanidoacétate méthyl
transférase.
Foie.
NH2
H2N+=C NH–CH2–COOH.
CH3.
ATP.
ADP
Créatine kinase.
NH~P
H2N+=C N–CH2–COOH.
CH3.
Muscle.
Créatine.
Créatine
phosphate.
Pi + H2O.
Cyclisation
spontanée.
O
NH–C
H2N+=C
N––CH2.
CH3.
Créatinine.
(éliminée dans les urines).
COOH
CH2
CH2
C=O
S~CoA
.
COOH
CH2–NH2.
Succinyl-CoA.
Glycine.
CoA-SH.
PPL.
ALA synthase mitochondriale.
(Etape limitante, régulée par l’hème).
COOH
CH2
CH2
C=O
CH–NH2
COOH
Acide α-amino-β-
cétoadipique
CO2.
ALA synthase
COOH
CH2
CH2
C=O
CH2
NH2
Acide δ-amino-
lévulinique (ALA).
COOH
CH2
CH2
C=O
CH2
NH2
COOH
CH2
CH2
C=O
CH2
NH2
2H2O.
ALA déshydratase
Cytoplasmique.
Mn2+.
COOH CH2
CH2 CH2
C–––––C
C CH
CH2 N
NH2 H
Porphobilinogène
(premier précurseur).
2 molécules de δ-
amino-lévulinate.

BiochAA. cours
Métabolisme des acides aminés – LEMAIRE.
5/32
Ceci est une dérivation du cycle de Krebs.
2.3.e) Les purines :
Glycine.
Glycine.
Guanine.
N
C
C
N
HC
NH2
N
CH
N
C
N
C
C
HN
C
O
N
CH
N
C
H2N
Adénine.
COOH
CH2
CH2
CHO
Semi-aldéhyde
succinique.
NAD+
NADH, H+
Déshydrogénase.
COOH
CH2
CH2
COOH
Succinate.
Succinyl-CoA.
Fumarate.
Oxaloacétate.
Citrate.
α-cétoglutarate.
COOH
CH2
CH2
C=O
COOH
Succinyl-CoA.
Glycine.
CO2.
CoA-SH.
ALA synthase.
δ-amino-
lévulinate.
α-cétoglutarate.
COOH
CH2
CH2
C=O
CHO
δ-semialdéhyde-lévulinate.
= α-cétoglutaraldéhyde.
FH4.
N5–N10–CH2–FH4.
NAD+
NADH, H+
Voie mineure.
Cycle
de
Krebs.
H2O.
Glycine.
Formyl-
transférase.
PPL
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
1
/
32
100%