L`électrophorèse

L’électrophorèse
SOMMAIRE :
Historique
Principes
Différents types d’électrophorèse
I) Electrophorèse sur papier et acétate de cellulose
II) électrophorèse sur gel
séparation sur gel de polyacrylamide (PAGE)
séparation sur SDS-PAGE
Electrophorèse bi-dimensionel avec deux dimensions (la première étant la
focalisation Isoélectrique (FIE) et la deuxième étant la SDS-PAGE)
séparation sur gel d’agarose
Electrophorèse en champ pulsé
III) Electrophorèse capillaire
IV) « Blotting » et applications
Western Blot
Séquençage d’ADN
Southern et Northern Blot
Zymographie
Cours effectué par Aude Barami (contact sur : aude@univ-st-etienne.fr (précisez que
l’on est étudiant en P1) ou à la porte 130 de la fac de médecine a bellevue)
INSERER LES SCHEMA QUI SERONT SUR LE BVRA

Historique
1937 : Tisetius met au point l’électrophorèse en veine liquide
1955 : Smithies utilise les gels d’amidon pour séparer les protéines sériques par
électrophorèse.
Sanger complète l’analyse de la séquence d’acides aminés de l’insuline bovine,
première protéine déchiffrée
1959 : Raymond et Weintroub introduisent les gels de polyacrylamide, meilleure séparation
des protéines par électrophorèse (au gel d’amidon).
1966 : Maizel introduit l’emploi du sodium dodécyl sulfate (SDS), perfectionne
l’électrophorèse des protéines sur gel de polyacrylamide.
1975 : O’ Farell développe les gels bi-dimensionnelle (focalisation isoélectrique, SDS-
PAGE), séparation de plus de 1000 protéines sur un même gel.
1984 : Schwartz et Cantor développent l’électrophorèse en champ pulsé : séparation de très
grosse molécules d’ADN
Principes
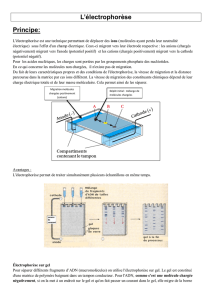
L’électrophorèse est une méthode de séparation des molécules chargées électriquement. On a
une séparation puis on a une caractérisation ou une purification de molécules d’intérêts. Le
terme « électrophorèse » vient de « electro » signifiant énergie électrique et de « Phoresis »
(photos en grec) qui veut dire porter, avoir en soi.
Cette méthode s’applique principalement à la biochimie et à la biologie moléculaire et on à
une séparation des protéines et des acides nucléiques.
Son principe consiste à avoir une migration de molécules chargées (perte de neutralité
électrique) dissoutes ou en suspension dans un solvant, sous l’effet d’un champ électrique.
Un champ électrique est produit par un générateur de courant continu. Le support du champ
est un tampon conduisant le courant d’un pôle à l’autre.
Les caractéristiques propres des molécules et les conditions de l’électrophorèse implique des
vitesse de migration des ions qui est différentes et une séparation les unes des autres (les
anioniques migrent vers l’anode (+) et les cationiques vers la cathode (-).
Les molécules à séparer sont déposées sur un support dont chaque extrémité est en contact
avec une solution tampon. Dans chaque solution tampon se trouve une électrode. Les
électrodes sont reliées à un générateur de courant.

Lorsque le générateur envoie du courant, les molécules chargées se déplacent sur le support
en direction de l’électrode de signe opposé à leur charge.
Différents types d’électrophorèse
On à plusieurs supports d’électrophorèse :
- sur supports liquide : « électrophorèse en veine liquide » pour les TRES grosses particules
(cellules, organites) qui est très peu utilisé.
- sur supports poreux : « électrophorèse de zone »
On à différents support d’électrophorèse de zones : - papier
- acétate de cellulose
- semi solide (gel)
Le support poreux doit être homogène et le plus inerte possible.
On à plusieurs types d’électrophorèse sur gel :
Electrophorèse sur gel d’agarose
Electrophorèse en champ pulsé
Electrophorèse sur gel de polyacrylamide (où PAGE pour polyacrylamide gel
electrophoersis)
Electrophorèse bi-dimensionnelle
Les électrophorèses peuvent aussi être réalisé en conditions dénaturantes (détergent types
SDS ou urée).
I) Electrophorèse sur papier et acétate de cellulose
On a une migration des molécules principalement en fonction (charge globale) en condition
non dénaturante.
L’électrophorèse sur papier :
On a une séparation des petites molécules (acides aminés ou petits peptides) puis une
détermination du point isoélectrique d’un acide aminé. On à une mesure de mobilité par
détermination de mobilité d’un acide aminé à différent pH et on a la relation suivante.

Mobilité= k . (pH-pHi) / Masse molaire
Cette méthode d’électrophorèse sur papier est peu utilisée de nos jours.
L’électrophorèse sur acétate de cellulose :
On a une séparation de molécules de milieux complexes (plasma), de petites molécules
migrant de manière proportionnel à leur charge.
Cette méthode peut être remplacé par des électrophorèses sur gel.
L’électrophorèse sur acétate de cellulose est fait de la manière suivante :
-imprégnation d’une bande par un électrolyte convenable
-dépôt d’une goutte de solution contenant l’espèce ionique à étudier sur la bande
-établissement d’un ddp entre les deux extrémités de la bande de papier
En milieu basique, les protéines sont chargées négativement. Dans le montage ci-dessus, on
a :
- les extrémités des bandes sont plongées dans des solutions conductrices de pH=9.2
- une application d’un champ électrique (entraînant la dissolution de l’échantillon dans la
bande conductrice et la migration le long de la bande).La vitesse de migration dépend de la
magnitude de la charge et de la taille des molécules.
- une coloration des protéines (rouge Ponceau)
- une analyse de la densité optique des bandes (l’intensité de la coloration est donc
proportionnelle à la concentration de protéines) et on obtient les spectres suivant :
Les espèces ioniques se déplacent
sous l’action d’un champ électrique
avec une vitesse propre

La vitesse de migration dépend de la magnitude de la charge. Chez des personnes atteint de
myélome, on à une immense concentration de γ-globuline.
II) Electrophorèse sur gel
Elle sépare des molécules selon leur ratio charge /masse.
Cette méthode conjugue l’effet de filtration du gel (taille des pores limitant la vitesse de
migration) et la mobilité électrophorétique. Les molécules atteignent rapidement une vitesse
V telle que :
V = q.E / f
q étant la charge de la particule, E étant le champ électrique, f étant le coefficient de
frottement de la particule par rapport à son solvant
D’après la théorie de l’électrophorèse, on à une mobilité U dans un champ électrique au sein
d’un gel
Log U = log U0 – Kr . C
Log U0 étant la mobilité en milieu liquide
Kr étant le coefficient de retardement dû au gel (fonction MW)
C qui est la concentration de gel
La vitesse de migration dépend aussi de la masse moléculaire.
On à deux types de matériaux pour l’électrophorèse sur gel :
- l’électrophorèse sur gel de polyacrylamide (l’acylamide à la formule suivante : CH2-CH-
CO-NH2 se polymérise avec le NN méthylène-bis-acrylamide et on à du polyacrylamide qui
à la formule brute suivante : CH2=CH-CO-NH-CH2-NH-CO-CH=CH2 qui donne des
chaînes latéral linéaire
- l’électrophorèse sur gel d’agarose qui utilise de l’agarose qui est un colloïde naturel extrait
d’algue rouge (Gracilaria) et est un polysaccharide gélifiant. On a dans ce cas des pores de
grande taille donc on sépare les très grosses molécules qui sont des protéines de plus de 500
kDa et des acides nucléique de plus de 1500 pb.
 6
7
8
9
10
11
12
13
14
15
16
6
7
8
9
10
11
12
13
14
15
16
1
/
16
100%