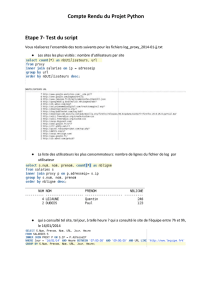
II. La structure électronique des atomes

CM11 - 1 -
I. Structure de l’atome
L’atome est le constituant élémentaire de la matière et il a une
structure discontinue. Il est formé d’un noyau central et entouré d’un nuage
électronique
1911 : Rutherford a placé une feuille d’or composé de 1000 couche d’atome
d’épaisseur ~ 5000 Â (=10-10 m) Il a bombardé cette cible par des atomes
d’hélium (2 protons / 2 neutrons). Derrière il a placé un écran fluorescent. Il
a remarqué 1 particule / 100 000 est dérivée : la matière est essentiellement
constituée de vide. On peut aussi conclure à l’existence d’un noyau
(dimension 10 000 fois plus petite que le l’atome).
La charge du noyau est positive (+ 1.6.10-14 C). Il est constitué de nucléons :
Protons
Nucléons
Le noyau occupe 1/1015 du volume total de l’atome. Son rayon est de 10-12
cm. Masse proton : mp =1.007276 u.m.a. (Unité de Masse Atomique)
=1.67239.10-27 kg
Masse neutron : mn =1.008665 u.m.a.
=1.6747. 10-27 kg
Masse électron : me =0.000549 u.m.a.
=9.108. 10-31 kg
Charge élémentaire d’un électron :
Qe- =-1.6.10-19 C
La masse d’un atome = masse du noyau
Le nombre de proton est égal au nombre d’électron car l’atome est
électriquement neutre.
II. Atome-gramme ; Masse atomique
Atome : particule très petite ~ 1Â
Pour travailler à l’échelle, on multiplie par N = 6.02.1023 (nombre
d’Avogadro)
Ex : X 10N taille d’un homme distance terre soleil
Proton et neutron 1/10 mm Ø / 1670 tonnes
e- balle 5 cm Ø / 900 kg
On travaille dans l’infiniment petit.
N atomes réels = 1 atome gramme = 1mole d’atome
npe mmm 1839
1
1836
1
1.

CM11 - 2 -
III. Molécule-gramme ; Masse molaire ; volume
molaire
Les atomes peuvent ainsi former une molécule.
N molécules réelles = 1 molécule gramme = 1 mole de molécule
Masse molaire : masse des atomes constitutifs de cette molécule.
Volume molaire : volume d’une mole de substance
Le volume molaire d’une substance gazeuse occupe dans les conditions
standards (0° / pression = 1 atmosphère) 22.4 l.
IV. Nombre de charges et nombre de masse
Nucléide ou nuclide :
Z : nombre de charges = numéro atomique
Fixe la position de l’élément dans le tableau périodique
A : nombre de masse = Z + n = p + n
Isotope d’un même élément = même nombre de protons mais nombre de
neutrons différents.
Ex : Le chlore naturel est formé de 2 isotopes
n = 18
n = 20
Pour définir la répartition des électrons autour du noyau, il existe deux
théories :
La mécanique classique
La mécanique quantique
I. Notions de modèles
a) La mecanique classique (BOHR –
SOMMERFELD)
Les électrons gravitent autour du noyau en décrivant des trajectoires
circulaires ou elliptiques.
1901 : Affirmation de PERRIN sur la trajectoire circulaire des e-.
1913 : mise en équation par BOHR
Postulat de BOHR : Le moment cinétique mvr ne peut prendre que les
valeurs discrètes égales à nh/2π
2h
nmvr
m : masse de e-
v : vitesse des e-
r : rayon de l’orbite
n : nombre quantique principal = nombre entier
h : constante de Planck = 6.62.10-34 J.s
Bien avant la théorie de BOHR, BALMER (1885) et LYMAN avaient
observé que les longueurs d’ondes de raies émises vérifiaient une relation
expérimentale généralisée par RITZ en 1908.
avec n2>n1
σ : nombre d’onde
λ : longueur d’onde de la radiation émise
RH : constante de RYDBERG = 1.0967776.107 m-1
Séries de raies selon les valeurs de n2 et n1
n1 = 1 n2 = 1, 2,3,… série de LYMAN UV
n1 = 2 n2 = 2,3, 4,… série de BALMER Visible
X
A
Z
Cl
Cl
37
17
35
17
2.
)
11
(
12
2
2
1nn
RH

CM11 - 3 -
n1 = 3 n2 = 3, 4,5,… série de PASHEN
n1 = 4 n2 =4, 5, 6… série de BRACKETT infra-
n1 =5 n2 =5, 6, 7,… série de PFIND rouge
Cependant on ne peut donner aucune explication pour de nouvelles raies, la
théorie de BOHR est insuffisante
.
1916 : amélioration de la théorie par l’ajout de trajectoires elliptiques
(SOMMERFELD) et en étendant la quantification de l’énergie.
Cette théorie reste inapte à expliquer le détail des spectres des
atomes à plusieurs électrons. Elle n’explique pas les trajectoires des atomes
lourds.
1920 : nouvelle approche des phénomènes mécaniques quantiques
b) La mecanique quantique ou ondulatoire
La notion de trajectoire ou d’orbite de l’e- perd toute signification :
L’hypothèse de DE BRÖGLIE (1924)
Tout électron en mouvement est associé à une onde caractérisée par λ :
mv
h
Principe d’incertitude d’HEINSBERG (1926)
On ne peut déterminer simultanément la position et la quantité de
mouvement de l’e-. Une grande précision sur l’une entraîne une grande
incertitude sur l’autre et inversement.
Position
Mouvement
Le mieux que l’on puisse faire dans ce cas c’est de déterminer la probabilité
de présence d’1 e- dans une position donnée (orbitale atomique).
Equation de SCHRÖDINGER (1926)
La probabilité de présence d’un électron peut s’exprimer à l’aide d’une
fonction d’onde ψ et qui représente l’amplitude associée à cette fonction
d’onde.
0)(
²
²8
e
hm
Δ : opérateur Laplacien
Ψ : amplitude associée à la longueur d’onde
E : énergie totale du système e- - noyau
Γ : énergie potentielle électrostatique du système e- - noyau
La résolution de cette équation permet de déterminer la signification de Ψ, n,
l et m.
c) Notion d’orbitale atomique (O.A.)
Dans la théorie de BOHR – SOMMERFELD, on a déterminé une onde de
l’électron par n, l, m ?
Une orbitale atomique est représentée par convention par une surface fermée
limitant le volume dans lequel la probabilité de présence de l’électron est de
95%. Forme de O.A. = f(l)
Orientation de O.A. = f(m)
m
h
vx
h
px
2
2

CM11 - 4 -
O.A. = (case quantique)
d) Les quatre nombres quantiques n, l, m, ms
Le nombre quantique principal n
n caractérise la couche et quantifie l’énergie.
n E [1 ;7]
n=1 couche K
n=2 couche L
…
n=7 couche Q
Le nombre quantique secondaire l
l caractérise la sous couche et décrit la forme orbitale et quantifie le moment
cinétique
l E [0 ; n-1]
l = 0 sous couche s (Sharp)
l = 1 sous couche p (principal)
l = 2 sous couche d (diffuse)
l = 3 sous couche f (fondamental)
Le nombre quantique magnétique m
m caractérise l’orbitale au case quantique et fixe son orientation dans un
champs magnétique
m E [-l ; +l] 2l + 1 valeurs
Le nombre quantique de spin ms = ± 1/2
Il correspond au centre de rotation de l’e- sur son axe et quantifie le moment
d’orientation.
Spin en haut +1/2 Spin en bas -1/2
N
L
M
Représentation
1
0
0
1s
2
0
0
2s
1
-1 / 0 / 1
2p
3
0
0
3s
1
-1 / 0 / 1
3p
2
-2 / -1 / 0 / 1 / 2
3d 3d
II. La structure électronique des atomes
Si un atome a Z électrons, il va être classé en tenant compte de trois règles :

CM11 - 5 -
a) La regle de KLECHKOWSKY
Les e- occupent à l’état fondamental des orbitales de plus basse énergie, ce
qui confère à l’atome une énergie minimum et une stabilité maximum. Le
remplissage se fait selon (n+l) croissant.
b) Principe d’exclusion de PAULI
Deux e- dans un atome ne peuvent avoir leur nombre quantique identique
(au moins un différent)
c) regle de HUND
p3 Etat excité
p3 Etat stable
Il faut commencer à remplir le maximum dans chacune des orbitales avant
de saturer.
d) Remarque
La sous-couche d est d’autant plus stable qu’elle est à moitié remplie ou
totalement remplie.
Ex : ns² (n-1)d4 ==< ns1 (n-1)d5
ns² (n-1)d9 ==< ns1 (n-1)d10
e) Exemple : Representation de la structure du
cuivre 29Cu
Représentation par des cases quantiques
Représentation par des symboles des niveaux d’énergie quantique
29Cu = 1s² 2s² 2p6 3s² 3p6 4s1 3d10
29Cu = [Ar] 4s1 3d10
Couche externe
Formulation / structure / configuration électronique d’un élément chimique
= OA vacante
e- célibataire
e- apparié
Remarque : Si tous les e- de la structure électronique sont appariés, la
structure est dite diamagnétique (≠ paramétrique). Les e- de la couche
externe sont dits e- de valence. Ce sont eux qui déterminent la réactivité de
l’élément.
III. Classification périodique des éléments
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
1
/
39
100%