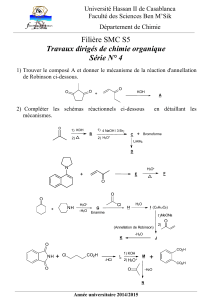
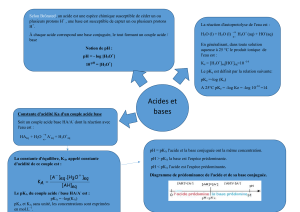
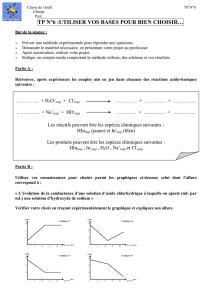
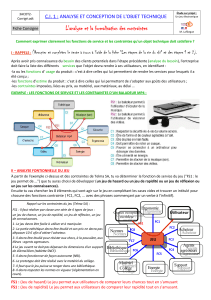
Royaume du Maroc
Université Hassan II
Faculté des sciences
Aïn Chock, Casablanca.
COURS DE REACTION
CHIMIQUE
Élaboré par : Professeur Abdelaziz EL AMRANI
CHIMIQUE

SOMMAIRE
CHAPITRE 1:
ÉQUILIBRES CHIMIQUES
CHAPITRE 2:
ÉQUILIBRES ACIDE
–
BASE
ÉQUILIBRES ACIDE
–
BASE
CHAPITRE 3:
EQUILIBRES DE DISSOLUTION-
PRECIPITATION
CHAPITRE 4:
EQUILIBRES D’OXYDO-REDUCTION

CHAPITRE I : EQUILIBRE CHIMIQUE
I - NOTION D'EQUILIBRE CHIMIQUE
1°) équilibre homogène
2°) équilibre hétérogène
3°) Ecriture du bilan de la réaction
II - LOI D'ACTION DE MASSE - CONSTANTE
D'EQUILIBRE
D'EQUILIBRE
1°) Expression de la constante d’équilibre
2°) Variation de Kp avec la température
III- LOIS DU DEPLACEMENT DE L’EQUILIBRE
1°) Modification de la température
2°) Modification de la pression
3°) Modification de la composition
4°) Variance d'un système

1. Introduction
Un état « d’équilibre chimique »
2 réactions de sens opposées
-Même vitesse
&
- Concentrations des réactifs et des
produits dans le milieu n’évoluent plus
avec le temps

Exemple :
CH3OH + CH3COOH CH3COOCH3+ H2O
Les deux systèmes (1) et (2) précédents évoluent l’un vers
(1)
Sens direct
(2)
Sens indirect
Méthanol acide acétique acétate de méthyle eau
Les deux systèmes (1) et (2) précédents évoluent l’un vers
l’autre et atteindront un état d’équilibre dynamique.
Les réactifs continuent de réagir. Il y a donc réactions directe
(de gauche à droite) et indirecte (de droite à gauche) de façon
continue.
il y a équilibre entre deux réactions. Les vitesses des deux
réactions étant identiques.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
1
/
192
100%