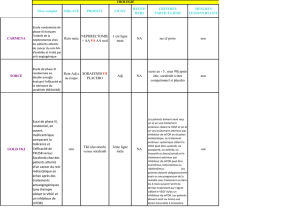
Mécanismes de résistance aux antiangiogéniques dans le cancer du rein Mechanisms of resistance to angio-angiogenics in renal cell carcinoma

Correspondances en Onco-urologie - Vol. II - n° 1 - janvier-février-mars 2011
28
dossier thématique
Rein :
traitements adjuvants
Mécanismes de résistance
aux antiangiogéniques
dans le cancer du rein
Mechanisms of resistance to angio-angiogenics in renal cell carcinoma
B. Beuselinck*, **, A. Karadimou**, ***, S. Oudard**, ***
* Oncologue médical,
hôpitaux universitaires
de Louvain, Belgique.
** Inserm U674 :
génomique fonctionnelle
des tumeurs solides,
Paris.
*** Service d’oncologie
médicale, hôpital européen
Georges-Pompidou, Paris.
L
e carcinome du rein représente 2 à 3 % de
tous les cancers dans le monde. Bien que la
majorité des malades soit guérie par néphrec-
tomie seule, jusqu’à un tiers des patients présentent
une maladie métastatique. Les nouvelles thérapies,
notamment les antiangiogéniques (comme le suni-
tinib, le sorafénib, le pazopanib et le bévacizumab,
ciblant le facteur de croissance endothélial vas-
culaire [VEGF]) et les molécules qui ciblent la voie
de la cible chez les mammifères de la rapamycine
(mTOR), tels l’évérolimus et le temsirolimus, ont
récemment remplacé les cytokines comme trai-
tements de première ligne dans le carcinome du
rein avancé ou métastatique. Le sunitinib, le bévaci-
zumab (combiné à l’interféron) et le pazopanib sont
les traitements de choix pour les carcinomes du rein
métastatiques de pronostic bon ou intermédiaire.
Le temsirolimus est un traitement possible en cas
de mauvais pronostic. Le sorafénib et l’évérolimus
sont actuellement utilisés comme traitements de
deuxième ligne. Parmi les antiangiogéniques, le
bévacizumab bloque le VEGF circulant, tandis que
le sunitinib, le sorafénib et le pazopanib sont des
inhibiteurs de la tyrosine kinase (ITK) du récepteur
du VEGF et du récepteur du Platelet-Derived Growth
Factor (PDGF). Le VEGF est le principal facteur pro-
angiogénique, et le PDGF intervient dans la conso-
lidation des nouveaux vaisseaux.
Bien que, avec ces nouvelles thérapies, nous obtenions
des réponses et des survies sans progression (SSP)
nettement meilleures qu’avec l’immunothérapie, cer-
tains malades (15 à 20 %) sont primairement résistants
à ces traitements (la résistance est en l’occurrence
primaire), et la maladie de la majorité de ceux chez
Points forts
highlights
»
La découverte de facteurs moléculaires génétiques prédictifs
de la réponse aux antiangiogéniques nous permettra à l’avenir
de sélectionner les malades candidats pour ces traitements de
façon à optimiser leur utilisation et à éviter les coûts inutiles et
les effets indésirables chez des malades qui n’en retireront pas
de bénéfice.
»
Nous disposons de données précliniques indiquant que la tumeur
pourrait mettre en route des voies alternatives d’angiogenèse
lorsque la voie principale de néo-angiogenèse, la voie du VEGF,
est bloquée.
»
Une autre possibilité consiste en une up-regulation de la voie VEGF,
de sorte que le blocage du VEGF ne soit plus suffisamment efficace.
»
La cellule tumorale pourrait également activer d’autres voies de
croissance moins dépendantes du métabolisme aérobique.
Mots-clés : Cancer du rein –Antiangiogéniques –Résistance.
The discovery of molecular-genetic factors predictive for
response on anti-angiogenics will enable us to optimalize
the selection of candidate patients for treatment. We will
be able to avoid unnecessary costs and side effects in some
patients.
Preclinical data are available on how tumors can activate
alternative angiogenic pathways when the principal
pro-angiogenic pathway, the VEGF-pathway, has been
blocked.
The upregulation of the VEGF-pathway, insufficiently
blocked by the anti-VEGF therapy, could be another
mechanism of resistance.
Finally, the tumor cell could activate alternative growth
pathways less dependant on aerobic metabolism.
Keywords: Kidney cancer –Antiangiogenics –Resistance.

Correspondances en Onco-urologie - Vol. II - n° 1 - janvier-février-mars 2011
29
Mécanismes de résistance aux antiangiogéniques dans le cancer du rein
qui une réponse partielle ou une stabilisation pro-
longée est obtenue nira tôt ou tard par progresser
(la résistance est alors secondaire).
Nous ne disposons pas de marqueurs prédictifs de
la réponse. Ces marqueurs seraient néanmoins très
utiles puisqu’ils nous permettraient de n’utiliser la
thérapie antiangiogénique que chez les patients
pouvant en bénéficier. Nous pourrions également
éviter effets indésirables et coûts en l’absence de
tout bénéfice.
Bien que nous ne disposions pas de marqueurs pré-
dictifs ables, plusieurs critères et classications pro-
nostiques de la SSP et de la survie globale (SG) ont été
développés.
La classication du Memorial Sloan-Kettering Cancer
Center (MSKCC) est la plus connue pour prédire la survie
en cas de cancer du rein localement avancé ou méta-
statique(1). Elle divise les malades en 3groupes de
risque (favorable, intermédiaire et risqué, défavorable),
selon 5facteurs associés à une survie moindre : le délai
entre le diagnostic et l’instauration du traitement sys-
témique (<1an), un taux élevé de LDH (>300 U/ ml) et
de calcium corrigé (>10 mg/ dl), un taux bas d’hémo-
globine (<11,5 g/ dl pour les femmes et <13 g/ dl pour
les hommes) et un score de Karnofsky bas (<80 %).
Ces critères, bien qu’ils aient été développés il y a une
dizaine d’années, au moment où l’immunothérapie
était le traitement de référence, ont été confirmés
récemment dans le cadre d’un essai pivotal évaluant
le sunitinib qui a inclus 375patients(2).
Dans cette étude, les facteurs prédictifs indépendants
associés à une SSP moindre sont un taux de LDH élevé,
la présence d’au moins 2sites métastatiques, l’absence
de néphrectomie, le performance status (PS) de l’Eas-
tern Cooperative Oncology Group (ECOG) [>0] et un
taux élevé de thrombocytes (> 400 000/ mm3). Notre
groupe a récemment publié une étude concernant
l’eet négatif sur la survie et sur la SSP de la présence
de métastases osseuses dans le cancer du rein traité
par sunitinib(3).
Comme nous pouvons le constater, il s’agit surtout
de marqueurs cliniques et biochimiques. En outre,
ils ne nous donnent pas susamment d’arguments
pour ne pas entamer de traitement chez un malade
donné. Peut-être trouverons-nous dans l’avenir des
critères plus ables au niveau moléculaire et géné-
tique. Jusqu’à aujourd’hui, cependant, aucun marqueur
prédictif moléculaire ou génétique de la réponse aux
antiangiogéniques n’a pu être décrit.
Nous passerons ici en revue les publications concer-
nant la résistance primaire et secondaire aux antiangio-
géniques.
Mécanismes d’action
des antiangiogéniques
Pour être en mesure de comprendre les mécanismes de
résistance aux antiangiogéniques, nous devons d’abord
comprendre leur mécanisme d’action. Les fonctions du
VEGF et du PDGF sont en eet nombreuses, et les cibles
des antiangiogéniques de type ITK, capables de bloquer
de nombreuses protéines, sont multiples. Cependant,
nous pensons que l’action principale des antiangio-
géniques consiste à stopper la néo-angiogenèse en
bloquant la fonction du VEGF. Ce concept se fonde
sur une augmentation importante de l’expression de
VEGF en cas de cancer du rein, à cause de l’inactivation
du gène von Hippel-Lindau (VHL). En raison de cette
inactivation, responsable de la métabolisation rapide
de l’Hypoxia-Induced Factor (HIF), ce dernier, un facteur
de transcription de nombreux gènes impliqués dans la
néo-angiogenèse, la promotion du cycle cellulaire et
la migration cellulaire, est activé. De nombreux essais
précliniques ont conrmé cette théorie(4-7). Le VEGF
stimule la formation de nouveaux vaisseaux par les
cellules endothéliales. Le PDGF est responsable de la
formation d’une couverture de péricytes autour des
cellules endothéliales consolidant le vaisseau nouvel-
lement formé. Le sorafénib a la particularité de bloquer
également le récepteur au Fibroblast Growth Factor,
FGFR-1.
Outre le blocage de l’apparition de nouveaux vaisseaux,
les antiangiogéniques pourraient également avoir des
eets immunomodulateurs, voire cytostatiques directs,
sur des cellules tumorales porteuses de récepteurs au
VEGF.
L’eet antiangiogénique semble cependant le plus
important eet antitumoral, ce que conrment des
techniques d’imagerie dynamique montrant le ux
sanguin dans les tumeurs.
Dans des modèles de xénogreffe de carcinome du
rein(7), des analyses par immuno-histochimie de
tumeurs réséquées après un traitement par sorafénib
montrent une diminution de l’importance de la micro-
vasculature et une nécrose. Un scanner de perfusion,
une résonance magnétique de perfusion et d’autres
techniques d’imagerie non invasives peuvent mettre
en évidence, peu de temps après l’instauration du trai-
tement, une diminution rapide et quasi complète du
ux sanguin témoignant de cette dévascularisation.
En revanche, au moment où la résistance au traitement
apparaît, ces mêmes techniques d’imagerie montrent
de façon cohérente une restauration du ux sanguin :
l’imagerie de perfusion révèle une restauration du ux
et l’immuno-histochimie une colonisation de la tumeur

Correspondances en Onco-urologie - Vol. II - n° 1 - janvier-février-mars 2011
30
dossier thématique
Rein :
traitements adjuvants
nécrotique par des cellules endothéliales. Ces données
suggèrent que le développement de la résistance ne
serait donc pas seulement un ajustement des voies de
signalisation intracellulaires de croissance tumorale,
mais plutôt un eort conjoint des cellules tumorales et
stromales pour rétablir une circulation qui sera moins
dépendante du VEGF, sans toutefois s’en affranchir
complètement.
Il a été observé, dans des cas de carcinome du rein
traités par PTK787/ZK222584, petite molécule inhibi-
trice du récepteur du VEGFR, que le ux sanguin tumoral
après 1mois de traitement, mis en évidence par IRM de
perfusion, était corrélé de façon signicative et posi-
tive avec des variations de la taille des tumeurs après
4mois de traitement(8). En outre, les changements de
perfusion dans la tumeur, mis en évidence après 1mois
de traitement (perfusion augmentée ou diminuée),
étaient plus prédictifs de la SSP que des critères mor-
phologiques tels que ceux du RECIST. En conclusion,
la persistance ou la nouvelle émergence de vaisseaux
semble donc liée à la résistance à ce type de traitement.
Résistance par augmentation de l’hypoxie
tumorale et de l’expression conséquente
de HIF-1α, VEGF et PDGF
La réapparition de vaisseaux dans la tumeur pourrait
être stimulée par l’hypoxie intratumorale, due à la
croissance tumorale, mais également à la disparition
des vaisseaux suite au traitement. L’hypoxie intratumo-
rale pourrait mener à la revascularisation du fait d’une
expression augmentée de HIF-1α et, en conséquence,
d’une expression augmentée de VEGF et PDGF.
Divers arguments semblent conrmer cette théorie.
Plusieurs études ont montré une augmentation du taux
circulant de VEGF et de PDGF en cours de thérapie par
antiangiogéniques bloquant le récepteur du VEGF. Dans
une étude de phase II avec le sunitinib, les diérences
du taux de VEGF (augmenté durant le traitement) et
du taux de récepteur soluble VEGFR-2 et-3 (diminué
durant le traitement) sont plus importantes chez les
patients ayant présenté une réponse partielle que chez
ceux n’en ayant pas présenté(9).
Dans l’étude d’enregistrement du sorafénib, une aug-
mentation du taux de VEGF circulant avait été observée
chez les patients traités par sorafénib, mais pas dans le
bras placebo. Ces taux élevés de VEGF sous traitement
antiangiogénique pourraient expliquer les phénomènes
de are-up ou de progression rapide observés à l’arrêt
des antiangiogéniques, voire pendant les 2semaines
d’interruption du sunitinib(10).
En outre, dans plusieurs études cliniques de phaseII,
nous avons pu constater une ecacité certaine, bien que
moins importante, du blocage de la voie de signalisation
VEGF lors d’une deuxième ligne d’antiangiogéniques,
après le développement d’une résistance à une première
ligne de thérapie anti-VEGF. Ces données suggèrent
que la néo-angiogenèse après blocage du VEGF reste
en partie dépendante du VEGF. Une étude de phaseII
sur le sunitinib, portant sur 62malades réfractaires au
bévacizumab, a rapporté un taux de réponse RECIST de
23 % et une SSP médiane de 7,1mois. De même, une
étude de phaseII avec l’axitinib, portant sur 62malades
réfractaires au sorafénib, a montré un taux de réponse
RECIST de 23 % et une SSP médiane de 7,4mois. Le
niveau de sensibilité de la tumeur à la deuxième ligne
de traitement pourrait dépendre de la puissance relative
de chaque molécule pour bloquer le récepteur VEGF.
Résistance par l’activation
de voies alternatives de néo-angiogenèse
La néo-angiogenèse développée au moment de la résis-
tance pourrait également être le résultat de la mise
en route de voies alternatives d’angio genèse indé-
pendantes du VEGF. En fait, en inhibant le VEGF et le
PDGF, nous n’inhibons qu’une partie des mécanismes
enclenchés par HIF. Un grand nombre de gènes sont
exprimés sous l’inuence de HIF, et l’inhibition du VEGF
et du PDGF pourrait même stimuler des mécanismes
compensatoires de survie.
Angiogenèse alternative
par le Fibroblast Growth Factor
Lorsque des souris atteintes de cancers neuroendo-
criniens pancréatiques sont traitées par des anticorps
monoclonaux anti-VEGFR-2, nous observons une
diminution initiale de 50 % de la taille de la tumeur et
de la densité de la microvasculature. Néanmoins, une
nouvelle croissance tumorale suivra malgré le fait que
le blocage anti-VEGFR était maintenu. Ce récepteur se
trouvait toujours sous forme déphosphorylée, mais
la RT-PCR, réalisée sur des cellules provenant d’îlots
résistants, pouvait mettre en évidence une augmenta-
tion de la transcription de plusieurs gènes de la famille
du Fibroblast Growth Factor (FGF), des éphrines et des
angiopoïétines(11). L’administration concomitante
d’un adénovirus codant pour une forme soluble du
récepteur du FGF-2 (qui arrive à lier plusieurs membres
de la famille FGF) diminue la croissance et la revascula-
risation. Ces expériences suggèrent que le FGF joue un
rôle important dans la croissance et la revascularisation

Correspondances en Onco-urologie - Vol. II - n° 1 - janvier-février-mars 2011
31
Mécanismes de résistance aux antiangiogéniques dans le cancer du rein
indépendante du VEGF. Notons aussi que l’interféronα
a une action inhibitrice du FGF(12). D’autres molécules
inhibitrices du FGF sont en cours de développement.
Néo-angiogenèse alternative par l’IL-8
Dans des cellules de cancer du côlon rendues dé-
cientes en facteurs de transcription HIF, la chémokine
IL-8 semble avoir un rôle dominant dans la généra-
tion et le maintien de la microcirculation tumorale.
Un anticorps anti-IL-8 était capable de bloquer la néo-
angiogenèse tumorale. L’IL-8 pourrait donc jouer un rôle
proangiogénique dans des situations où la voie VEGF
est déréglée (par des mutations) ou bloquée par des
traitements anti-VEGF(13).
Néo-angiogenèse alternative
par le Placental Growth Factor
Le Placental Growth Factor (PlGF) est un homologue du
VEGF qui intervient dans le switch angiogénique. Un
anticorps anti-PlGF est capable d’inhiber la croissance
et l’apparition de métastases dans plusieurs tumeurs,
y compris dans celles qui résistent aux inhibiteurs du
récepteur du VEGF(14). En outre, les taux plasmatiques
de PlGF sont augmentés chez les malades atteints
d’un carcinome du rein et recevant du sunitinib(15).
Cependant, le sunitinib bloque le VEGFR1, qui est aussi
le récepteur du PlGF. Le blocage du PlGF ne serait donc
probablement pas un traitement actif en cas de résis-
tance aux anti-VEGF dans le carcinome du rein.
Néo-angiogenèse alternative
par le blocage de la voie Tie-2/Ang-2
L’axe formé par l’angiopoïétine2 (Ang-2) et son récep-
teur Tie-2 semble avoir de nombreuses fonctions, parmi
lesquelles la néo-angiogenèse en parallèle avec l’axe
du VEGF(16). Dans des études précliniques, l’inhibi-
tion de Ang-2 a mené à la suppression de la crois-
sance tumorale(17). L’Ang-2 stimule la production de
métallo protéinases matricielles (MMP) via les récepteurs
des intégrines(18). Les MMP stimulent à leur tour la
sécrétion du VEGF(19). Les MMP servent à détruire la
membrane basale, nécessaire à la néovascularisation.
En outre, les taux plasmatiques d’Ang-2 semblent aug-
menter chez les patients traités par sunitinib. L’inhibition
d’Ang-2 pourrait donc freiner ou prévenir la néovas-
cularisation en cas de résistance aux traitements ciblant
le VEGF. L’AMG-386, une immunoglobuline liée au
récepteur Tie-2, est capable de bloquer la xation de
l’Ang-2 à son récepteur. La combinaison d’AMG-386
et de sorafénib a permis un taux de réponse de 29 %
chez des malades atteints d’un carcinome du rein, dont
certains étaient résistants aux anti-VEGF(20).
Pour nir, notons que J.Garcia-Donas(21) a présenté,
à l’ESMO2010, un abstract montrant une associa-
tion statis tiquement signicative entre 1SNP (Single
Nucleotide Polymorphism) dans le VEGF et 2SNP dans le
VEGFR-3 et le temps jusqu’à progression sous sunitinib :
les malades dont le système VEGF fonctionne mal répon-
dent moins bien au traitement anti-VEGF, peut-être
parce que l’angiogenèse tumorale repose davantage
sur d’autres mécanismes proangiogéniques chez eux.
Résistance par l’activation
de voies alternatives de croissance
An d’assurer leurs possibilités de croissance, les cellules
tumorales ont besoin de nouveaux vaisseaux, mais elles
mettent aussi en œuvre un ensemble de mécanismes
de croissance qui impliquent des récepteurs de surface
suivis de cascades intracytoplasmiques, elles-mêmes
aboutissant à l’activation de facteurs de transcription
nucléaires et à l’expression de nombreux gènes. Les
voies de transmission intracellulaires sont diciles,
mais le complexe de mTOR semble jouer un rôle central.
Cette voie est très souvent activée dans les cellules
du cancer du rein, bien que ce ne soit jamais de façon
constitutionnelle. Ainsi, les inhibiteurs de la voie mTOR,
tels que l’évérolimus, ont pu prouver une ecacité,
même modeste, dans le cancer du rein résistant aux
traitements antiangiogéniques. Dans l’étude RECORD-1,
portant sur 410malades atteints d’un cancer du rein
réfractaire aux anti-VEGF, la SSP était de 4,0mois sous
évérolimus, contre 1,9mois sous placebo. Le taux de
réponse objective n’était cependant que de 1 %.
J.L.Perez-Gracia et al. ont collecté du sérum lors de
l’instauration du traitement et au moment de l’évalua-
tion de la réponse, réalisée par RECIST chez 31patients
traités par sunitinib(22). Le sérum de 6patients avec
des phénotypes extrêmes de réponse claire (3patients)
ou de progression rapide (3patients) a été analysé :
174cytokines impliquées dans l’angiogenèse et la pro-
lifération tumorale ont été recherchées. Les taux des
cytokines des 2groupes ont été comparés. Vingt-sept
des 174cytokines variaient de façon signicative selon
que les patients avaient présenté une réponse partielle
ou une progression de la maladie. Les 6cytokines les
plus pertinentes du point de vue statistique et biolo-
gique (TNFα, MMP-9, ICAM-1, BDNF, SDF-1α et VEGF)
ont été analysées chez 22malades évaluables, et les
résultats ont été corrélés au bénéce clinique (réponse
ou stabilisation de la maladie) ou à la progression. La
conclusion de J.L.Perez-Gracia etal. est que, dans le
sérum de malades atteints d’un cancer du rein méta-

Correspondances en Onco-urologie - Vol. II - n° 1 - janvier-février-mars 2011
32
dossier thématique
Rein :
traitements adjuvants
statique, le taux de TNFα et de MMP-9 à l’inclusion est
signicativement plus élevé chez les non-répondeurs
au sunitinib et qu’il est signicativement corrélé à la
SSP et à la SG.
Résistances primaire et secondaire
par inhibition inadéquate
du récepteur du VEGF
Des métastases de cancer du rein, comme la tumeur
primitive, pourraient progresser sous traitement anti-
VEGF du fait de l’augmentation de l’expression du récep-
teur ou à cause de taux plasmatiques insusants du
traitement anti-VEGF.
La probabilité d’obtenir un bénéce clinique est liée
de façon positive aux concentrations plasmatiques
du sunitinib et de l’axitinib (un ITK anti-VEGF en phase
de développement)[23]. Plusieurs publications ont
en fait montré un lien entre les eets indésirables du
traitement anti-VEGF (par exemple, l’hypertension pour
l’axitinib) et son ecacité. En outre, la dose d’ITK n’est
en général pas adaptée au poids du malade. Certains
sujets –ayant des taux plasmatiques insusants de
sunitinib, de sorafénib ou de pazopanib– sourent
moins des eets indésirables, mais l’ecacité théra-
peutique est alors également moindre. Le changement
des traitements pour les comorbidités des patients
pourrait interagir avec la métabolisation du sunitinib
via le cytochromeP450.
Une étude randomisée de phase II avec le sorafénib a
montré que l’escalade de la dose de sorafénib jusqu’à
600mg×2/j a pu induire une diminution de la taille
tumorale chez 42 % des patients, alors que la maladie
progressait lorsque la posologie était de 400mg× 2/ j.
Bien que des réponses selon RECIST n’aient pas pu être
obtenues, la SSP médiane en cas d’escalade de dose
était de 3,6mois. Cette SSP médiane correspond à celle
obtenue avec l’évérolimus en phase de progression sous
ITK anti-VEGF et est donc supérieure à celle observée
sous placebo(24).
Statut VHL et résistance :
aucune corrélation ?
Étant donné le rôle important joué par les mutations
du gène VHL dans la pathogenèse du carcinome du
rein, plusieurs groupes ont étudié la corrélation entre
le statut VHL et la réponse aux antiangiogéniques.
L’hypothèse était que l’absence de protéine VHL indui-
rait des taux plus élevés de HIF et une angiogenèse plus
importante. L’inhibition de la voie du VEGF serait donc
plus ecace dans les carcinomes du rein porteurs de
la mutation de VHL ou ayant des gènes promoteurs
hyperméthylés.
Vingt-six (60 %) des 43patients observés par B.I.Rini
et al.(25) étaient porteurs de mutations de VHL ou
atteints d’hyperméthylation du promoteur ; 17 (40 %)
étaient VHL sauvage. Les taux de réponse aux antiangio-
géniques (interféronα +bévacizumab, sunitinib ou
axitinib) étaient respectivement de 48 % et 35 %, avec
un temps médian jusqu’à progression de 10,8mois
versus 5,5mois. La diérence du temps médian jusqu’à
progression n’était pas signicative (p=0,26), mais le
devenait (p=0,06) lorsque les 15malades porteurs
d’une hyperméthylation VHL ou d’une mutation VHL
tronquante étaient comparés aux malades n’ayant pas
ces anomalies génétiques : le temps médian jusqu’à pro-
gression était alors de 13,3mois versus 7,4mois). B.I. Rini
et al. ont conclu que les malades atteints d’un cancer
du rein métastatique porteurs d’hyper méthylation de
VHL ou de mutations tronquantes avaient un temps
jusqu’à progression prolongé sous traitement anti-
angio génique.
T.K. Choueiri a refait le même exercice sur 123patients
atteints d’un carcinome du rein à cellules claires méta-
statiques traités par sunitinib, sorafénib, axitinib ou
bévacizumab(26). Les patients atteints d’une inacti-
vation du gène VHL ont mieux répondu au traitement
que les patients VHL sauvage (les taux de réponse
globale étaient de 41 % versus 31 % [p=0,34]). Le
taux de réponse était encore plus important (51 %)
chez les malades porteurs d’une mutation non-sens.
Une analyse multivariée comprenant plusieurs autres
facteurs pronostiques cliniques importants a montré
que la présence d’une mutation avec perte de fonction
reste un facteur pronostique indépendant associé à une
meilleure réponse. Cependant, la SSP et la SG n’étaient
pas aectées par ces mutations. Les patients avec une
mutation de VHL avaient une SSP médiane de 12,0mois
versus 9,0mois en cas de wild-type et de 11mois en
cas de VHL hyperméthylé (p=0,78). La SSP médiane
des malades ayant une mutation de VHL avec perte
de fonction était de 13,7mois versus 9,0mois chez les
malades VHL sauvage (p=0,71).
La résistance secondaire
ne serait probablement pas induite
par des mutations secondaires
La thérapie antiangiogénique exerce son eet sur les
cellules endothéliales vasculaires, et donc plutôt sur
 6
6
1
/
6
100%