D É d i t o r i a l Du neuf

Éditorial
191
Act. Méd. Int. - Métabolismes - Hormones - Nutrition, Volume VI, n° 5, septembre-octobre 2002
écrite par Addison en 1855, l’insuffisance surrénale (IS) primaire a
toujours fasciné par ses signes cliniques spectaculaires, son évolution
mortelle brutalement transformée, au siècle précédent, par les corti-
coïdes, qui en font aujourd’hui, en exagérant un peu, un défaut presque
anodin, car facile à substituer et compatible avec une vie normale. Nous
évoquons souvent le président Kennedy auprès de nos patients, parfois
flattés et toujours rassurés par cet exemple, malgré son destin tragique.
Encore plus spectaculaire, l’insuffisance surrénale aiguë reste un exemple
mythique d’urgence médicale rare qui préoccupe l’interne de garde, tout
autant par sa crainte de méconnaître le diagnostic que de l’évoquer à tort.
Pour le diagnostic biologique de l’IS, que de progrès accomplis depuis le
test à l’eau de Robinson, le test de Thorn comptabilisant les éosinophiles
sous ACTH et le dosage des 17-cétostéroïdes (premier dosage hormonal) !
Ces étapes historiques, résultant d’une démarche intelligente, ont fait
place à la biologie moderne, facilement accessible sur quelques micro-
litres de sang, et démonstrative de l’IS, dont l’origine haute ou basse est
clairement démarquée.
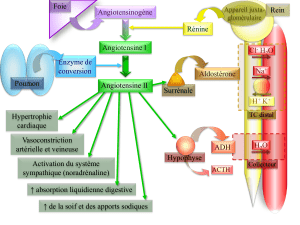
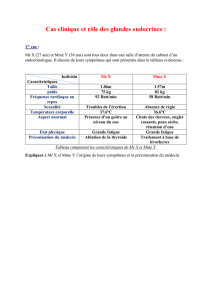
Dans ce numéro, M. Quinkler et ses collaborateurs font une très bonne
mise au point diagnostique et thérapeutique de l’IS en 2002. La clinique
n’a pas changé, mais l’expérience et les statistiques ont introduit un
meilleur relief dans la sémiologie clinique et surtout biologique. On sait,
par exemple, que le test au Synacthène®ne départage pas bien les addi-
sonniens des insuffisants hypophysaires et on en comprend la raison : le
récepteur de l’ACTH doit être soumis à une stimulation régulière par
l’ACTH (endogène ou exogène) pour stimuler la synthèse de cortisol. La
simple confrontation du taux de cortisol et d’ACTH est en général suffi-
sante pour le diagnostic de l’IS et du niveau lésionnel.
Le traitement s’affine et, depuis quelques années, les doses substitutives
d’hydrocortisone sont revues à la baisse : progressivement, les endocri-
nologues français rabattent leurs traditionnels 30 mg/j à 20 mg/j ou même
15 mg/j, comme leurs voisins européens. Cependant, la substitution cor-
ticoïde n’est probablement pas parfaite, faute de critères de surveillance
assez précis, surtout dans l’IS d’origine haute. La place de la DHEA, qui
paraît logique dans l’IS après la ménopause, reste à confirmer.
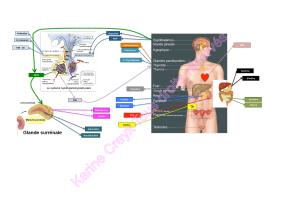
C’est surtout dans l’étiologie de l’insuffisance surrénale que les dernières
années ont apporté du neuf. La tuberculose n’est plus la première cause
Du neuf
avec du vieux
J. Mahoudeau*
* Service d’endocrinologie, CHU de Caen.
D

Act. Méd. Int. - Métabolismes - Hormones - Nutrition, Volume VI, n° 5, septembre-octobre 2002
Éditorial
192
de maladie d’Addison. La surrénale a rejoint la thyroïde et le pancréas
parmi les cibles de l’auto-immunité. La poly-endocrinopathie auto-
immune de type 1, associant des déficits endocriniens multiples, dont
l’IS et l’hypoparathyroïdie, et une candidose par déficit immunitaire,
a trouvé son support génétique dans les altérations du gène AIRE
(autoimmune regulator), dont on a décrit 45 mutations sans corrélation
phénotype/génotype. L’excellente revue de H. Lefebvre souligne l’intérêt
théorique et aussi pratique de cette avancée.
L’IS de l’enfant est surtout représentée par le déficit en 21-hydroxylase,
dont la fréquence justifie le dépistage néonatal systématique en France.
La revue de G. Pinto-Primard et M. Polak indique là aussi des progrès
exponentiels : la génétique est connue, la démarche diagnostique et thé-
rapeutique est claire, le traitement anténatal prévient la virilisation du
fœtus féminin. Les causes rares d’IS de l’enfant ont ouvert des voies qui
éclairent les mécanismes génétiques du développement et de la trophi-
cité du cortex surrénal, notamment les adrénoleucodystrophies, les
mutations inactivatrices du récepteur de l’ACTH, le syndrome des 3 A et,
surtout, les anomalies des gènes SF1 (steroidogenic factor 1) et DAX1
(dosage-sensitive sex-reversal, adrenal hypoplasia congenita, X chromo-
some). SF1 est un facteur de transcription de la stéroïdogenèse et on
comprend que les mutations de ce gène provoquent une IS et un hypo-
gonadisme. On comprend moins comment les mutations de DAX1
entraînent ces mêmes pathologies (et un déficit gonadotrope) puisque,
normalement, DAX1 réprime SF1. Ces pathologies d’exception sont,
comme souvent, d’un grand enseignement, et le cortex surrénal reste la
cible d’une recherche intense.
En pratique, le diagnostic d’une IS, indépendamment de sa cause, haute
ou basse, est d’une importance vitale et, aujourd’hui, il est facile le plus
souvent. Il ne reste plus qu’à espérer la possibilité d’un diagnostic en
temps réel, que les endocrinologues envient à d’autres spécialistes.
Disposera-t-on jamais de dosages instantanés du cortisol et de l’ACTH ?
En attendant ce jour, la clinique reste reine et permet de prendre une
décision thérapeutique rapide en attendant les résultats des dosages.
1
/
2
100%