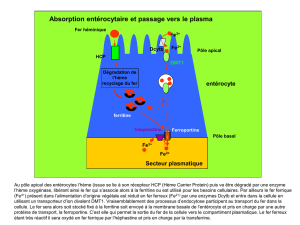
facteurs erythropose bichat2009-2

Y
Y
Pool de fer
libre
Y
YY
Y
Y
Y
Récepteurs à la transferrine
Y
Y
Fe
2+
Apo-IRP [4Fe-4S]-IRP
Hétéropolymères de ferritine
Y
Y
endosome
Apo-Tf
DMT1
H+V-ATPase
3+
Fe -Transferrine - 3+
Fe
Capture, transport et stockage du fer dans la cellule
Capture, transport et stockage du fer dans la cellule
Mitochondrie
Endocytose?

Globules rouges
Foie
Macrophage
(rate, moelle, foie)
Hème oxygénase
Fe(II)
Céruloplasmine Fe(II)
Fe(III)
Fe(III)-Tf
Ferroportine
Apo-Tf
HFE
Hepcidine
Recyclage du fer par les macrophages

MV
ON
H
M P P M M V
C
HN
H
C
H2
N
HC
HN
H
O
NADPH + H+
NADP+
Biliverdine réductase A
(BVRA)
Bilirubine
MV
ON
H
M P P M M V
C
H
NC
H
N
HC
HN
H
O
+ CO
Hème
NADPH + H+
H2O + NADP
Fe2+
Hème Oxygénase
(HO)
Biliverdine
O2
Cytochrome P450 réductase
Recyclage Macrophagique

Hepcidine et HFE régulent l’absorption intestinale du fer
et les taux de fer des macrophages
Absorption du fer
Recyclage du fer
Entérocyte mature Macrophage
Hepcidine
XX
HFE

Hepcidine est un senseur des réserves en fer
Expression activée par la
surcharge en fer
Déficit complet en hepcidine
Surcharge en fer tissulaire
(foie, pancréas, cœur)
Déplétion des réserves en fer
macrophagiques
Store regulator
Réprime l ’absorption intestinale du Fer
Bloque la sortie cellulaire du fer
par interaction avec la ferroportine
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
1
/
56
100%