I. Influence des états physiologiques

Pharmacologie
LES FACTEURS DE VARIATIONS DE L’ACTIVITE DES MEDICAMENTS
- La réponse au médicament dépend de :
o Pathologies associées : rénale, cardiaque, respiratoire, etc.
o Age.
o Sexe.
o Etat nutritionnel.
o Adhésion au traitement.
o Interactions médicamenteuses.
o Génétique.
I. Influence des états pathologiques
II. Influence des états physiologiques
III. Influence du polymorphisme génétique
IV. Autres facteurs de variation
I. Influence des états physiologiques
- Deux grandes pathologies modifient la pharmacocinétique des médicaments :
o Insuffisance rénale.
o Insuffisance hépatique.
Insuffisance rénale
- La fonction rénale estimée par la clairance à la créatinine selon la formule de Cokcroft.
- CLcréatinine = (140−Age)x poids
crétinémie x 1,03 (femme)
ou x 1,24 (homme)
- Une insuffisance rénale chronique doit être définie par la clairance à la créatinine :
o <60 mL.min-1 chez l’adulte.
o < 50 mL.min-1 chez la personne âgée.
- Lors d’une insuffisance rénale il y a deux modifications dans la pharmacocinétique :
o Diminution de fixation aux protéines plasmatiques.
o Diminution de l’élimination urinaire et filtration glomérulaire.
- Il faut donc une adaptation posologique pour les médicaments fortement éliminés par le rein :
o Diminuer la dose en conservant le rythme d’administration.
o Augmenter l’intervalle d’administration de la même dose.
Insuffisance hépatique
- Elle est plus dure à quantifier.
- Les modifications induites sont complexes.
1. Variations d’ordre pharmacocinétique
Au niveau de la résorption
- Vitesse de vidange gastrique :
o Accélération entraine une résorption plus rapide.
o Ralentissement (Parkinson) favorise la dégradation de la molécule dans l’estomac.
- Diminution de la mobilité intestinale (post-op) diffère de la résorption.
- Maladies inflammatoires du tube digestif (maladie de Crown).
Au niveau de la distribution
- Hypo-albuminémies majeures modifient la fixation plasmatique.
- Exemple, insuffisance rénale : augmentation fraction libre multiplié par 2 pour la phénytoïne et
par 6 pour la coumadine.
- Troubles acido-basiques affectent pénétration tissulaire.

Au niveau de la métabolisation
- Les produits sont très dépendants du débit sanguin hépatique (variations hémodynamiques).
- Produits à clairance hépatique faible (sensibilité à l’altération de l’activité enzymatique).
Au niveau de l’excrétion rénale
- Accumulation si diminution de la filtration glomérulaire.
- Compétition des « métabolites urémiques » et des médicaments au niveau de la sécrétion
tubulaire.
2. Variations d’ordre pharmacodynamique
- Les récepteurs, cibles des médicaments sont des éléments régulés.
- La variation du nombre de récepteur : cause majeure de changement de réponse de l’organisme à
un médicament.
- Désensibilisation : diminution de la réactivité cellulaire à un médiateur ou à une molécule
exogène.
- L’hypersensibilité (augmentation de la réactivité).
- Désensibilisation et hypersensibilité sont des phénomènes expliquant les phénomènes de
tolérance et les syndromes de sevrage.
Exemple : malade parkinsonien sont traités par dopathérapie (administration de L-DOPA). Après
3 à 5 ans de dopathérapie il y a une perte d’efficacité et des effets indésirables (modifications de
la sensibilité des récepteurs dopaminergiques).
II. Influence des états physiologiques
1. L’enfant
- L’enfant n’est pas un adulte en miniature : les paramètres pharmacocinétiques de nombreux
médicaments modifiés chez l’enfant.
- 1ère année : maturation de la résorption.
o Nouveau né : motilité intestinale, pH gastrique, etc. instables = influence de la résorption
des médicaments par voies orales.
o Nourrisson et enfant : résorption accélérée.
- Diminution de la fixation protéique (rôle de la bilirubine, albumine fœtale, etc.) = fraction du
médicament liée aux protéines est abaissée (valeur adultes atteintes vers 10-12 mois).
Au niveau de la métabolisation
- Activité des enzymes microsomiales est faible : absence de glucuroconjugaison
(chloramphénicol) = altération du métabolisme de nombreux médicaments.
Exemple : transformation théophylline en caféine surtout chez le prématuré (surveillance taux de
théophylline et de caféine).
- 1ère année : maturation de l’élimination jusqu’à 6-8 mois fonction rénale réduite avec
déséquilibre entre fonction glomérulaire et tubulaires.
2. La personne âgée
- Avec l’âge association pathologies rénales hépatiques et cardiaques.
Au niveau de la résorption
- Moins bonne résorption : pas de conséquences thérapeutiques notables.
- [protéines plasmatiques]sang diminue un peu : effet mineur.
Au niveau de la métabolisation
- Métabolisme hépatique modifié :
o Métabolisme oxydatif et de l’hydroxylation diminuent avec l’âge.
o Pas de modifications des mécanismes de conjugaisons (acéthylation, glucuronidation).
Au niveau de l’excrétion rénale
- Baisse de l’élimination rénale : favorise l’accumulation des médicaments et les El doses-
dépendants.

III. Influence du polymorphisme génétique
1. Variations d’ordre pharmacocinétique
Au niveau de la métabolisation
- Le polymorphisme d’acétylation (isoniazide). Malades réagissant de façons différentes au
traitement par Isoniazide : RIMIFON.
- Dans une population, répartition bimodale de la capacité d’acétylation définissant les acétylateurs
lents (demi-vie d’environ 6h) et rapide. La répartition varie selon les ethnies :
POPULATION
% acétylateurs rapides
Caucasiens
30 à 50
Africains
40-60
Asiatiques
90-95
- Le caractère acétylateur lent se transmet selon el mode autosomique récessif.
- A posologie standar les uns résistent au traitement els autres présentent des accdides toxiques.
- Pour une posologie standard les risques d’apparition d’efferts indésirables sont différents :
o Acétylateurs lents : effets II neurologiques augmentent.
o Acétylateurs rapides : la production plus importante, in situ, d’un métabolite
hépatotoxique majorerait le risque d’hépatite.
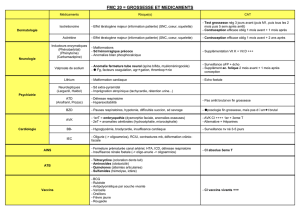
MEDICAMENTS, GROSSESSE ET ALLAITEMENT
- La prescription médicamenteuse chez la femme enceinte nécessite une évaluation du bénéfice
thérapeutique attendu par la mère au regard du risques connus (ou potentiels) pour le futur enfant
en fonction de son âge gestationnel au moment du traitement.
- Toutes les femmes potentiellement enceintes et enfants systématiquement sont écartés des essais
cliniques.
- Ethiquement inadmissibles d’exposer le fœtus aux risques liés à l’absorption dont les
conséquences sont incertaines, si l’état de la mère ne l’impose pas.
- Etymologie de tératogène :
o Teras, teratos signifiant monstre en grec.
o Genos signifiant origine en grec.
Se dit de toute substance pouvant provoquer un développement anormal de l’embryon et
conduisant par là même à des malformations.
- Distinction de périodes au cours de la grossesse, toxicité différente selon la période :
o Après l’implantation : loi du tout ou rien, soit mort fœtal soit aucune incidence.
o Stade embryonnaire : période de l’organogénèse, risque tératologique majeur, à l’origine
de malformations.
o Après le 3ème mois : risque d’intoxication fœtal
- Les organes les plus précocement sensibles aux effets tératogènes sont :
o Le système nerveux central.
o Le cœur.
o Puis les membres, les yeux, les oreilles, et les organes génitaux externes.
- Principal danger : médicaments absorbés par automédication. Les patientes enceintes ne doivent
pas prendre de médicaments sans avis médical préalable.
- Difficultés de prévoir la tératogénicité d’un médicament car il n’y a pas d’essais thérapeutiques
chez la femme enceinte.
o Les données d’observations (pharmacovigilance) sont donc acquises par expérimentation
animale sur 3 espèces différentes dont une n’est pas un rongeur. Il y a donc des difficultés
d’obtenir des données de tératogénèse chez l’homme qui repose sur des cas rencontrés
o Intérêt de la déclaration aux centres de pharmacovigilance : constitution de référence.
o Informations limitées dans les RCP.
Particularités pharmacocinétiques au cours de la grossesse
- Modifications de l’absorption dues à :

o La diminution de la sécrétion de l’acide gastrique (40%), augmentation du pH gastrique.
o La diminution de la motilité intestinale.
- Au cours de la distribution :
o Augmentation albuminémie maternelle.
o Augmentation de la distribution, l’existence d’un placenta et du fœtus fait que le
médicament trouve un nouveau site de distribution.
Médicaments tératogènes
Thalidomide
- Effets nocifs pour le fœtus illustrés par la dramatique affaire du thalidomide (immuno-
modulateur utilisé comme hypnotique et antiémétique pour combattre les nausées matinales).
- Il a été utilisé de 1957 à 1962 : 12000 cas de malformations recensés (absence d’oreille interne,
paralysie des nerfs crâniens, phocomélies, etc.).
- C’est un inhibiteur de l’angiogenèse, interférant avec le développement des vaisseaux sanguins
surtout en cas de prise au cours des 25 à 50 premiers jours de la grossesse.
- Essais menés chez l’animal en 1954 pas de mise en évidence de la toxicité particulière. Idem
pour les essais thérapeutiques.
- Contre-indication absolue pendant la grossesse.
Distilbene
- Diéthylstilbestrol Distilbene prescrit dans ales années 70 pur prévenir les fausses couches =
80000 filles exposées pendant la grossesse.
- Anomalies génitales féminines / hypofertilité / augmentation cancer du vagin et du col de l’utérus
1977 prescription pendant la grossesse interdite.
Autres médicaments à risque tératogène important
Anti-acnéiques rétinoïdes : Roaccutane et génériques
- Hautement tératogène : SNC, cœur, oreilles, interruption de grossesse.
- Prescription :
o Action prolongée imposant contraceptions très stricte 1 mois avant la prescription.
o Test mensuels de la grossesse négative poursuivie 1 mois après l’arrêt.
Dérivé de la vitamine A, Etrétinate (Soriatane) : traitement de dermatoses
graves et psoriasis
- Hautement tératogène : SNC, squelette, Interruption de grossesse.
- Prescription : action prolongée imposant contraception très stricte moins avant le début du
traitement. Médicaments anti- cancéreux
- Tous les médicaments cytotoxiques ou antimitotiques présentent un risque tératogène majeur
s’ils ne provoquent pas la mort du fœtus.
- Si exposition à un traitement chimio-thérapeutique anticancéreux avant la conception risque de
malformations graves et fréquentes (25%).
Quelques Classes Thérapeutiques
Antidiabétiques
- Hyperglycémie est tératogène.
- Les antidiabétiques oraux (ex : Metformine, glucophage) sont contre-indiqués pendant la
grossesse.
- L’insuline est le traitement de choix + contrôle strict de la glycémie.
Anti-Inflammatoires Non-Stéroïdiens (AINS)
- AINS contre indiqué au cours de la grossesse :
o Risque de fermeture prématurée du canal artériel.
o Toxicité rénale.
o Risque hémorragique.
- Attention à l’automédication par AINS (migraine, lombalgie, etc.).
- Aspirine mêmes règles que pour les AINS sauf prescription faible dose (100mg par jour)
traitement de l’hypertension gravidique.

Traitement de la douleur
- Molécule antalgique à utiliser pendant la grossesse avec précaution : paracétamol.
- Attention à la confusion paracétamol aspirine en automédication.
Traitement hypertenseur
- IEC : contre indiqué anurie fœtale.
- ARA 2 : contre indiqués.
- Antagonistes calciques : contre indiqués.
- Bétabloquants : en second intention risque d’hypotension, hypoglycémie à la naissance relais
par un autre antihypertenseur.
Lithium
- Thymo-régulateur.
- Malformation cardiaque dans environ 4 à 8 % des cas.
- La lithémie doit être < 1meq/L.
- Diagnostic anténatal par équipe spécialisée en écho cardiaque fœtale à partir de la 20ème semaine.
AVK
- Héparine ne traverse pas le placenta et n’a aucun effet tératogène / pas excrété dans le lait
maternel.
- AVK traversent la barrière placentaire et sont excrétés dans le lait maternel tératogènes avec un
risque précoce (<9sem) : 5% des malformations.
Antiépileptiques
- Les crises convulsives font courir un grave danger à la mère et à l’enfant : indispensable de
traiter les femmes enceintes.
- Tous les antiépileptiques traversent la barrière fœto-placentaire + augmentation risque
malformations congénitale : fente labiale, malformations cardiovasculaires.
Anti rétroviraux
- Traitement par AZT rétroviraux de la mère HIV + césarienne.
- Diminution du taux de transmission materno-fœtale du virus HIV.
Médicaments et allaitement
- Rechercher des informations sur les risques.
- Décision au cas par cas.
- Evaluer le passage dans le lait.
Conclusion :
- L’utilisation des médicaments durant la grossesse et l’allaitement doit être surveillée.
- Peu d’informations sur les RCP.
- Evaluation en fonction des patients.
ANTAGONISTES CALCIQUES
I. Introduction
- Classe pharmacologique importante utilisée dans l’HTA et l’insuffisance coronaire.
- Blocage canaux calciques voltage dépendants de type L (de type lent).
- Trois groupes :
o Dihydropyridines.
o Phényl-alkylamines : Verapamil Isoptine®.
o Benzothiazine : Diltiazem Tildiem®.
II. Mécanismes d’action
- Inhibition du transfert membranaire de calcium dans les cellules musculaire cardiaques et les
cellules musculaires vasculaires.
- Diminue les résistances périphériques vasculaires.
- Et diminue la consommation en oxygène du myocarde.
- Mécanismes majeurs de régulation de la concentration cytosolique de calcium.
- SCHEMA
 6
7
8
9
6
7
8
9
1
/
9
100%