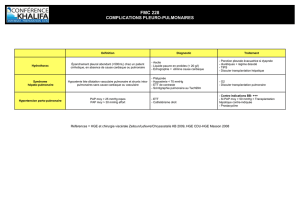
Lire l`article complet

169
La Lettre du Pneumologue - Vol. II - n° 5 - octobre 1999
u cours de la cirrhose, la mise en évidence d’une
hypoxémie est fréquente. Elle peut relever de
multiples causes, comme une pathologie bron-
chopulmonaire ou cardiaque associée, ou plus rarement d’ano-
malies vasculaires pulmonaires liées à l’hypertension portale,
s’intégrant dans le cadre d’un syndrome hépatopulmonaire
(SHP). Porter le diagnostic de SHP est important, car sa sévé-
rité intervient dans l’indication de transplantation hépatique.
DÉFINITION
Le SHP associe une hypertension portale, une augmentation du
gradient alvéolo-artériel d’oxygène (O2) et des dilatations vas-
culaires pulmonaires. L’hypertension portale est le plus sou-
vent secondaire à une cirrhose. La fréquence du SHP au cours
de la cirrhose (environ 20 %) est corrélée à la sévérité de
l’hépatopathie (1). Les mécanismes des anomalies vasculaires
pulmonaires du SHP, multifactoriels et de pathogénie com-
plexe (2, 3), sont dominés par une vasodilatation pulmonaire,
potentiellement réversible. Leur connaissance guide le choix
des examens complémentaires et du traitement (2, 4).
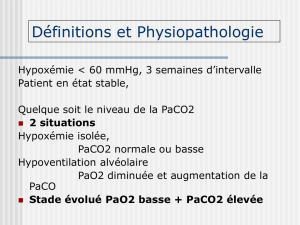
PHYSIOPATHOLOGIE DE L’HYPOXÉMIE
Différents mécanismes sont à l’origine d’une augmentation du
gradient alvéolo-artériel d’O2: hétérogénéité des rapports ven-
tilation-perfusion ( ), shunts et troubles de la diffusion
(1, 2, 3). Leur importance respective a été précisée par la tech-
nique d’élimination des gaz inertes.
MISE AU POINT
Le syndrome hépato-pulmonaire
●F. Chabot*, V. Robert*, J.M. Polu*
A
* Service des maladies respiratoires et réanimation respiratoire, CHU Nancy-
Brabois, Vandœuvre-lès-Nancy.
POINTS FORTS
POINTS FORTS
●
Le syndrome hépato-pulmonaire (SHP) est défini par une triade associant hypertension portale, augmentation du gra-
dient alvéolo-artériel d’oxygène et dilatations vasculaires pulmonaires dont la pathogénie serait liée à un déséquilibre
entre facteurs vasodilatateurs et vasoconstricteurs.
●
L’hypoxémie est multifactorielle avec une hétérogénéité régionale des rapports ventilation/perfusion, parfois des shunts
intrapulmonaires et un défaut de “diffusion-perfusion”.
●
Les signes évocateurs de SHP sont une dyspnée d’effort, une hypoxémie avec alcalose ventilatoire, une platypnée avec
orthodéoxie, parfois un trouble de diffusion sans syndrome obstructif ni restrictif. L’échocardiographie de contraste est
l’examen de choix pour le diagnostic des dilatations vasculaires.
●
L’inefficacité des traitements médicaux fait discuter la transplantation hépatique en cas d’hypoxémie sévère (PaO2infé-
rieure à 50 mmHg) améliorée sous 100 % d’O2(PaO2supérieure à 400 mmHg).
L’hétérogénéité des rapports est secondaire à une
inadaptation des débits sanguins à la ventilation régionale, liée
à une anomalie du tonus vasculaire due essentiellement aux
dilatations vasculaires intrapulmonaires (2). L’hypoxémie qui
en résulte est habituellement modérée, grâce à l’effet protecteur
de facteurs extrapulmonaires (déplacement vers la droite de la
courbe de dissociation de l’hémoglobine, augmentation du
débit cardiaque et de la ventilation minute).
Les shunts. Les shunts porto-pulmonaires et pleuro-pulmo-
naires sont rares et ont un retentissement fonctionnel négli-
geable par rapport à celui des shunts intrapulmonaires.
Expérimentalement, il existe une dilatation extrême des capil-
laires pulmonaires et, dans une moindre mesure, une néoangio-
genèse (5), mais la mise en évidence anatomique de shunts
intrapulmonaires est exceptionnelle chez l’homme. Sur le plan
morphologique, l’angiographie pulmonaire permet d’individua-
liser deux types de lésions, des dilatations artériolaires distales
diffuses (type 1 selon Krowka) et/ou exceptionnellement des
anastomoses artério-veineuses localisées, de gros calibre (type
2) (2). La technique d’élimination des gaz inertes, réservée à
quelques centres, est la méthode de référence pour évaluer le
retentissement fonctionnel. En pratique clinique, la quantifica-
tion des shunts est obtenue par la scintigraphie ou, mieux, par
le cathétérisme cardiaque droit réalisé sous 100 % d’O2.
L’hypoxémie induite par des shunts intrapulmonaires est plus
sévère que celle due à l’inhomogénéité des rapports . En
fait, ces deux mécanismes sont souvent intriqués, parfois asso-
ciés à des anomalies évoquant des troubles de la diffusion (2,
3). La coexistence d’anomalies vasculaires et d’altérations du
transfert du CO a conduit au concept de “défaut de diffusion-
perfusion” (2). Les dilatations vasculaires augmentent la dis-

tance de diffusion de l’oxygène entre l’alvéole et le centre du
capillaire. En air ambiant, la PO2alvéolaire (PAO2) est estimée
à 150 mmHg (figure 1). Le gradient de pression alvéolo-capil-
laire n’est pas suffisant pour permettre une diffusion de l’oxy-
gène jusqu’au centre du capillaire et donc pour oxygéner com-
plètement le sang veineux mêlé. L’hypoxémie est accentuée
par l’augmentation du débit cardiaque, caractéristique du syn-
drome hyperkinétique de la cirrhose, qui diminue le temps de
contact capillaire. Sous 100 % d’O2, la PAO2est supérieure à
600 mmHg et deviendrait suffisante pour permettre aux molé-
cules d’O2d’atteindre le centre du capillaire. La réponse à
l’oxygène dépendrait de l’étendue des dilatations vasculaires
(2). L’orthodéoxie, ou baisse de la PaO2lors du passage en
orthostatisme, est due à la répartition préférentielle du débit
sanguin dans les vaisseaux dilatés, prédominant aux bases pul-
monaires (2).
Au total, l’hypoxémie du SHP associe à des degrés divers une
hétérogénéité régionale des rapports , des shunts intra-
pulmonaires et un défaut de “diffusion-perfusion” (2, 3).
MÉCANISMES DES ANOMALIES VASCULAIRES
PULMONAIRES
La réversibilité fréquente des anomalies du tonus vasculaire
après transplantation hépatique suggère un trouble fonctionnel
(5, 6). Le mécanisme de la perte du tonus vasculaire pulmo-
naire est inconnu, peut-être lié à un excès de production de
médiateurs vasodilatateurs ou de facteurs angiogéniques (6), à
un défaut d’épuration hépatique d’un vasodilatateur pulmo-
naire ou à une métabolisation hépatique de vasoconstricteurs.
Le rôle de la prostacycline, suspecté expérimentalement, n’a
pas été confirmé chez l’homme (2).
Le NO a un rôle majeur dans la vasodilatation du SHP. Il est
synthétisé par l’intermédiaire de la NO synthétase (NOS). Il
existe plusieurs isoenzymes de la NOS, dont la NOS induc-
tible, ou iNOS, et la NOS constitutive, ou cNOS. L’endotoxi-
némie, fréquente au cours de la cirrhose, active l’iNOS, et
favoriserait une synthèse accrue de NO qui pourrait être à
l’origine de la vasodilatation générale avec état hyperkinétique
observée au cours de la cirrhose. La compréhension de la
pathogénie des dilatations vasculaires intrapulmonaires a été
facilitée par la mise au point d’un modèle expérimental de
SHP. Dans ce modèle, il existe une baisse de la vasoconstric-
tion à la phényléphrine. Cette dernière est restaurée par un pré-
traitement avec un inhibiteur de la NOS, suggérant que l’hypo-
réactivité vasculaire pulmonaire à la phényléphrine résulte
d’une augmentation de la production de NO (7). Plusieurs
observations sont en faveur d’une hyperproduction de NO
chez l’homme également. La concentration de NO exhalé au
cours du SHP est corrélée au gradient alvéolo-artériel d’O2.
Chez un patient atteint de SHP, la PaO2et le taux de NO expiré
se sont normalisés après transplantation hépatique. Dans un
autre cas, l’administration intraveineuse de bleu de méthylène,
un oxydant qui bloque l’action du NO, a corrigé transitoire-
ment l’hypoxémie et le shunt, en position couchée et assise.
Le rôle d’autres médiateurs vasoactifs ou angiogéniques
comme l’endothéline 1 est vraisemblable. Les constatations
rapportées lors du traitement de certaines cardiopathies congé-
nitales suggèrent qu’un facteur hépatique présent normalement
dans le sang veineux hépatique intervient dans l’angiogenèse
pulmonaire (6).
MANIFESTATIONS CLINIQUES
Les signes respiratoires évocateurs de SHP sont une dyspnée
d’effort et une platypnée, qui est une aggravation de la dys-
pnée lors du passage en orthostatisme. Un hippocratisme digi-
tal et une cyanose sont parfois observés quand l’hypoxémie est
sévère.
Dans 80 % des cas, les symptômes respiratoires sont décou-
verts chez un patient présentant une hypertension portale ou
une hépatopathie connues. Les angiomes stellaires sont parti-
culièrement fréquents.
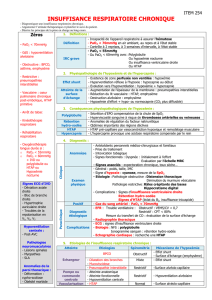
EXAMENS COMPLÉMENTAIRES
(tableau I)
L’hypoxémie est associée à une alcalose ventilatoire chronique
et fréquemment à une baisse du coefficient de transfert du CO,
sans trouble ventilatoire obstructif ni restrictif. L’orthodéoxie
est très évocatrice d’un SHP. L’hypoxémie, aggravée par
l’effort, s’améliore sous 100 % d’O2en l’absence de malfor-
mations artério-veineuses de gros calibre (2).
La radiographie thoracique, habituellement normale, peut
mettre en évidence un syndrome interstitiel basal minime lié à
une augmentation de la trame vasculaire pulmonaire, ou
exceptionnellement des fistules artério-veineuses (6).
La dilatation des vaisseaux intrapulmonaires est constante au
cours du SHP. La scintigraphie de perfusion permet d’affirmer
la présence d’un shunt (sans en préciser le siège) par la détec-
tion d’une radioactivité extrapulmonaire (2). L’échocardio-
graphie de contraste est la technique de référence dans le
dépistage des shunts droits-gauches (2). Les microbulles,
injectées dans une veine périphérique, sont normalement
MISE AU POINT
170
La Lettre du Pneumologue - Vol. II - n° 5 - octobre 1999
Alvéole
PaO2 = 90
Capillaire normal
PAO2 = 150 mmHg
Dilatation capillaire
PAO2 = 150 mmHg
Dilatation capillaire
PAO2 = 650 mmHg
Alvéole
Diamètre
capillaire
8-15 µ
15-100 µPaO2 < 60 PaO2 > 500
Alvéole
{
Figure 1. Concept de “diffusion-perfusion”.
Le gradient de pression alvéolo-artériel d’O2(PAO2–PaO2) est représenté
par les flèches. En air ambiant, il est insuffisant pour permettre une dif-
fusion de l’oxygène jusqu’au centre du capillaire dilaté et pour oxygéner
normalement le sang veineux. Sous 100 % d’O2, il permet aux molécules
d’oxygène d’atteindre le centre du capillaire.
Adapté de Lange et Stoller (2).

171
La Lettre du Pneumologue - Vol. II - n° 5 - octobre 1999
absorbées lors de leur passage pulmonaire. La distinction entre
shunt intracardiaque et shunt intrapulmonaire est basée sur leur
délai d’apparition dans les cavités cardiaques gauches. Dans
les shunts intracardiaques, les microbulles apparaissent un à
trois battements après leur passage dans les cavités droites ; un
délai de quatre à six systoles suggère l’existence d’un shunt
intrapulmonaire. L’utilisation de la voie transœsophagienne
sensibilise la technique et précise la provenance des micro-
bulles (habituellement veines lobaires inférieures). Une écho-
cardiographie de contraste normale exclut quasiment le dia-
gnostic de SHP (2).
Le développement de l’échocardiographie a réduit les indica-
tions de l’angiographie pulmonaire, méthode invasive qui indi-
vidualise des lésions de type 1 et/ou exceptionnellement de
type 2 (2), qui peuvent aussi être mises en évidence par
l’angioscanner.
TRAITEMENT
La valeur de la PaO2, en air ambiant et sous 100 % d’O2, guide
les choix thérapeutiques (4) (tableau I). L’oxygénothérapie a
une efficacité variable selon l’importance des lésions vascu-
laires. La persistance d’une hypoxémie sévère sous O2traduit
habituellement un shunt important. Les données physiopatho-
logiques, encore incomplètes, ne permettent pas actuellement
de proposer un traitement pharmacologique efficace.
L’objectif est une correction de l’hypoxémie par la restaura-
tion d’un tonus vasculaire pulmonaire normal avec l’utilisation
d’antagonistes de vasodilatateurs, de vasoconstricteurs pulmo-
naires, ou en diminuant les taux plasmatiques de médiateurs
présents en excès au cours du SHP (6). Il n’y a pas d’améliora-
tion significative avec les inhibiteurs des prostaglandines
(indométacine), les analogues de la somatostatine, le bismési-
late d’almitrine, les sympathomimétiques (isoprénaline) ou les
bêtabloquants (propranolol), les antagonistes des estrogènes
(tamoxifène) ou les échanges plasmatiques (2). Dans un cas de
lymphadénopathie immuno-angioblastique, le SHP a guéri
après traitement étiologique de la maladie auto-immune (8).
Toutefois, l’amélioration d’un SHP sous traitement médical est
exceptionnelle (2).
La pose d’un shunt portosystémique intrahépatique (TIPS)
a été exceptionnellement associée à la régression d’une
hypoxémie sévère, mais ce succès, inexpliqué, n’a pas été
confirmé (9).
L’angiographie pulmonaire permet la mise en évidence et
l’embolisation de fistules artérioveineuses. Elle est proposée
quand la PaO2est inférieure à 150 mmHg sous 100 % d’O2, car
elle révèle parfois des fistules artérioveineuses qui peuvent
être embolisées (4).
La transplantation hépatique corrige parfois l’hypoxémie (4,
10). Le SHP peut être une indication de transplantation hépa-
tique en cas d’hypoxémie sévère, inférieure à 50 mmHg (2, 4,
10). La mortalité postopératoire est corrélée à la sévérité de
l’hypoxémie (4, 10). Le taux de succès de la transplantation
hépatique est plus élevé quand la PaO2préopératoire est supé-
rieure à 400 mmHg sous 100 % d’O2, valeur seuil proposée
pour retenir l’indication d’une transplantation hépatique (4).
L’intérêt de la transplantation hépatique n’est pas établi chez
les patients très hypoxémiques, avec une faible réponse sous
100 % d’O2. Dans cette situation, l’indication est discutée au
cas par cas selon les résultats de l’angiographie (4).
CONCLUSION
Au cours du SHP, les dilatations vasculaires pulmonaires sont
à l’origine d’une hypoxémie dont la sévérité peut altérer le
pronostic vital. L’hypoxémie peut être une indication de trans-
plantation hépatique, seul traitement efficace actuellement. ■
RÉFÉRENCES BIBLIOGRAPHIQUES
1. Moller S., Hillinger J. et coll. Arterial hypoxaemia in cirrhosis : fact or fic-
tion ? Gut 1998 ; 42 : 868-74.
2. Lange P.A., Stoller J.K. The hepatopulmonary syndrome. Ann Intern Med
1995 ; 122 : 521-9.
3. Robert V., Chabot F. et coll. Syndrome hépatopulmonaire : physiopathologie
des anomalies des échanges gazeux. Rev Mal Respir 1999 : sous presse.
4. Krowka M.J., Porayko M.K. et coll. Hepatopulmonary syndrome with pro-
gressive hypoxemia as an indication for liver transplantation : case reports and
literature review. Mayo Clin Proc 1997 ; 72 : 44-53.
5. Schraufnagel D.E., Malik R. et coll. Lung capillary changes in hepatic cir-
rhosis in rats. Am J Physiol 1997 ; 272 : L139-L147.
6. Hervé P., Lebrec D. et coll. Pulmonary vascular disorders in portal hyper-
tension. Eur Respir J 1988 ; 11 : 1153-66.
7. Chabot F., Mestiri H. et coll. Role of NO in the pulmonary artery hyporeacti-
vity to phenylephrine in experimental biliary cirrhosis. Eur Respir J 1996 ; 9 :
560-4.
8. Cadranel J.L., Milleron B.J. et coll. Severe hypoxemia-associated intrapul-
monary shunt in a patient with chronic liver disease : improvement after medi-
cal treatment. Am Rev Respir Dis 1992 ; 146 : 526-7.
9. Corley D.A., Scharschmidt B. et coll. Lack of efficacy of TIPS for hepatopul-
monary syndrome. Gastroenterology 1997 ; 113 : 728-30.
10. Battaglia S.E., Pretto J.J. et coll. Resolution of gas exchange abnormalities
and intrapulmonary shunting following liver transplantation. Hepatology 1997 ;
25 : 1228-32.
Contexte hypertension portale, dyspnée d’effort
PaO2hypoxémie, orthodéoxie fréquente
RP normale habituellement
EFR volumes et débits normaux, TCO abaissé
Scintigraphie de perfusion shunt D-G (fixation extrapulmonaire)
Échocardiographie de contraste shunt D-G intrapulmonaire
PaO2(100 % O2)> 400 mmHg ➪transplantation hépatique ?
< 150 mmHg ➪angiographie pulmonaire
➪embolisation si fistule artérioveineuse
Tableau I. Diagnostic et traitement du SHP.
ANNONCEURS
BRISTOL-MYERS SQUIBB (Taxol), p. 166 ; RHÔNE-POULENC RORER (Institutionnelle, Nasacort), p. 168, p. 172 ;
PROMEDICA (Beclojet 250), p. 183 ; GÉNOPHARM (Logecine), p. 177 ;
GLAXOWELLCOME (Flixotide, Serevent), p. 186, p. 187 ; PASTEUR VACCINS (Pneumo 23), p. 203 ; SYNTHÉLABO (Mizollen), p. 204.
1
/
3
100%