Graphique 1

UNIVERSITESIDIMOHAMMEDBENABDELLAH
FACULTEDEMEDECINEETDEPHARMACIE
FES
AnnéeThèseN°/201006210
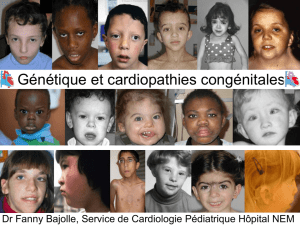
PROFILEPIDEMIOLOGIQUEETETIOLOGIQUE
DESCARDIOPATHIESCONGENITALES
(Etuderétrospectiveàproposde427cas)
THESE
PRESENTEEETSOUTENUEPUBLIQUEMENTLE07/04/2010
PAR
Néele04Février1985àOujda
Mlle.AKKAROUAFAE
POURL'OBTENTIONDUDOCTORATENMEDECINE
MOTS-CLES:
Cardiopathiescongénitales-Epidémiologie-Etiologies
JURY
M. Professeur
M. Professeur
M. Professeur
M. Professeur
M. Professeur
HIDAMOUSTAPHA.......................................................
dePédiatrie
ATMANISAMIR.............................................................
agrégédePédiatrie
BOUHARROUABDELHAK..............................................
dePédiatrie
AKOUDADHAFID........................................................
agrégédeCardiologie
HARANDOUMUSTAPHA...............................................
agrégéd’Anesthésieréanimation
JUGES
PRESIDENT
UNIVERSITESIDIMOHAMMED
BENABDELLAH
FES
RAPPORTEUR

1
LISTE DES ABREVIATIONS
AD : Autosomique dominant
Ao : Aorte.
AP : Atrésie pulmonaire.
APSO : Atrésie pulmonaire à septum ouvert.
AR : Autosomique récessif.
AT : Atrésie tricuspide.
ATS : Antithyroïdien de synthèse.
BAV : Bloc auriculoventriculaire.
BSA : Bloc sino auriculaire.
CAV : Canal atrioventriculaire.
CC : Cardiopathie congénitale.
CCM : Cardiopathie congénitale malformative.
CHU : Centre Hospitalier Universitaire.
CIA : Communication interauriculaire.
CIV : Communication interventriculaire.
CM : Cardiomyopathie.
CMD : Cardiomyopathie dilatée.
CMH : Cardiomyopathie hypertrophique.
CMR : Cardiomyopathie restrictive.
CMV : Cytomégalovirus.
COA : Coarctation de l’aorte.
DS : Déviation Standard.
ESA : Extrasystole auriculaire.
ESV : Extrasystole ventriculaire.
FC : Fausse couche.
FEE : Fibroélastose endocardique
HCD : Hypoplasie du cœur droit.
HCG : Hypoplasie du cœur gauche
HTA : Hypertension artérielle.
IAA : Interruption de l’arche aortique.
IAo : Insuffisance aortique.
IM : Insuffisance mitrale.
IP : Insuffisance pulmonaire.

2
IT : Insuffisance tricuspide.
J : Jour.
MFIU : Mort fœtale in utéro.
MPS : Mucopolysaccharidose.
Nbr : Nombre.
OD : Oreillette droite.
OG : Oreillette gauche.
PCA : Persistance du canal artériel.
PDVM : Prolapsus de la valve mitrale.
RAA : Rhumatisme articulaire aigu.
RAO : Rétrécissement aortique.
RASV : Rétrécissement aortique supra valvulaire.
RPM : Retard psychomoteur.
RSP : Retard staturo- pondéral.
RVPAP : Retour veineux pulmonaire anormal partiel.
RVPAT : Retour veineux pulmonaire anormal total.
SNC : Système nerveux central
SP : Sténose pulmonaire.
SPP : Sténose pulmonaire périphérique.
STB : Sclérose Tubéreuse de Bourneville.
STT : Syndrome transfuseur transfusé.
TAC : Tronc artériel commun.
TC : Tachycardie.
TGVx : Transposition des gros vaisseaux.
T21 : Trisomie 21.
T4F : Tétralogie de Fallot.
VCI : Veine cave inférieure
VCS : Veine cave supérieure.
VD : Ventricule droit.
VDDI : Ventricule droit à double issu.
VG : Ventricule gauche.
VGDI : Ventricule gauche à double issu.
VU : Ventricule unique.
WPW : Wolff Parkinson White.
XR : Récessif lié à X.

3
PLAN
INTRODUCTION .................................................................. 9
PARTIE THEORIQUE ............................................................11
I) Rappel sur l’embryologie cardiaque ............................................... 12
II) Adaptation à la vie extra utérine. .................................................. 22
III) Classification des Cardiopathies Congénitales ............................. 26
1) Classification anatomique .......................................................... 26
2) Classification embryologique ..................................................... 29
3) Classification physiopathologique ............................................. 32
IV) Diagnostic des Cardiopathies Congénitales ................................. 35
V) Traitement des Cardiopathies Congénitales ................................. 53
PARTIE PRATIQUE ..............................................................55
MATERIEL D’ETUDE ........................................................................... 56
RESULTATS DE LA SERIE HOSPITALIERE .................................................... 57
I) EPIDEMIOLOGIE ........................................................................................... 57
A) REPARTITION GEOGRAPHIQUE ..................................................................... 57
B) NIVEAU SOCIO ECONOMIQUE ....................................................................... 57
C) FREQUENCE................................................................................................. 58
1) REPARTITION GLOBALE ............................................................................ 58
a) Fréquence des cardiopathies congénitales ............................................ 58
a-1) L’incidence hospitalière .............................................................. 58
a-2) L’incidence saisonnière ............................................................... 58
b) Répartition selon l’âge ......................................................................... 60
c) Répartition selon le sexe ...................................................................... 62
2) REPARTITION SELON LE TYPE DE CARDIOPATHIES CONGENITALES ............ 63
a) Cardiopathies congénitales malformatives .......................................... 63
a-1) Les shunts gauche-droite : ............................................................... 63
1) CIV ............................................................................................... 63
a) fréquence ................................................................................... 63
b) âge ............................................................................................ 63
c) sexe ........................................................................................... 65
d) lésions associées ........................................................................ 65
2) CIA ............................................................................................... 67
a) fréquence ................................................................................... 67

4
b) âge ............................................................................................ 67
c) sexe ........................................................................................... 68
d) lésions associées ........................................................................ 68
3) PCA ............................................................................................... 69
a) fréquence ................................................................................... 69
b) âge ............................................................................................ 69
c) sexe ........................................................................................... 71
d) lésions associées ........................................................................ 71
4) CAV .............................................................................................. 71
a) fréquence ................................................................................... 71
b) âge ............................................................................................ 72
c) sexe ........................................................................................... 73
d) lésions associées ........................................................................ 73
a-2) Les cardiopathies cyanogènes ......................................................... 74
1) TGVx ............................................................................................. 74
a) fréquence ................................................................................... 74
b) âge ............................................................................................ 74
c) sexe ........................................................................................... 76
d) lésions associées ........................................................................ 76
2) T4F ................................................................................................ 77
a) fréquence ................................................................................... 77
b) âge ............................................................................................ 77
c) sexe ........................................................................................... 78
d) lésions associées ........................................................................ 78
3) RVPAT ........................................................................................... 79
a) fréquence ................................................................................... 79
b) âge ............................................................................................ 79
c) sexe ........................................................................................... 79
d) lésions associées ........................................................................ 80
4) VDDI ............................................................................................. 80
a) fréquence ................................................................................... 80
b) âge ............................................................................................ 80
c) sexe ........................................................................................... 80
d) lésions associées ........................................................................ 81
5) APSO ............................................................................................. 81
a) fréquence ................................................................................... 81
b) âge ............................................................................................ 81
c) sexe ........................................................................................... 82
d) lésions associées ........................................................................ 82
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
1
/
191
100%