République Algérienne Démocratique et Populaire
Ministère de l’Enseignement Supérieur et de la Recherche Scientifique
Université Mouloud MAMMERI Tizi-Ouzou
Faculté de Médecine
Sclérose en plaques
Conférence de quatrième année Médecine
Module de Neurologie / Neurochirurgie
Dr M. LOUNIS
Université Mouloud MAMMERI Tizi-Ouzou
Mise à jour: Octobre 2020

SOMMAIRE
1. INTRODUCTION
2. EPIDEMIOLOGIE
3. ÉTIOPATHOGENIE
3.1 ETIOLOGIES
3.1.1. SUSCEPTIBILITE GENETIQUE
3.1.2. FACTEURS ENVIRONNEMENTAUX
3.2. PHYSIOPATHOLOGIE
4. CLINIQUE
4.1. MANIFESTATIONS CLINIQUES
4.1.1 Mode de début
4.1.2 Phase d’état
4.2 FORMES EVOLUTIVES
5. ÉLEMENTS DU DIAGNOSTIC
5. 1 DIAGNOSTIC POSITIF
5.1.1 Imagerie
5.1.2 Électrophysiologie
5.1.3 Biologie
5.1.4 Critères diagnostiques
5.2 DIAGNOSTIC DIFFERENTIEL
6. TRAITEMENTS
6.1 TRAITEMENT DE LA POUSSEE
6.2 TRAITEMENT DE FOND
6.3 TRAITEMENT SYMPTOMATIQUE

1. INTRODUCTION
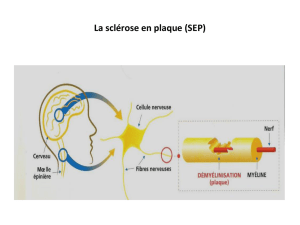
La sclérose en plaques (SEP) est définie comme une maladie inflammatoire démyélénisante du systme
nerveux central (cerveau et moelle épinire). Maladie auto-immune associée à un phénomne
neurodégénératif. Elle est caractérisée par la formation de « plaques » de sclérose sur la myéline, d’o
son appellation. La SEP est à l’origine de déficit moteurs et sensitifs, visuels ou cognitifs, de troubles
de l’équilibre, de douleurs...et résultent de la variation de la localisation des plaques. Elle évolue par
épisodes aigus imprévisibles, appelés poussées, puis, d’une manire générale évolue vers un mode
progressif.
La prise en charge de la SEP s’attache à traiter les poussées lorsqu’elles surviennent, à en prévenir leurs
survenues, à ralentir la progression du handicap, et à soulager les différents troubles associés.
2. EPIDEMIOLOGIE
Figure n°1: Prévalence de la SEP dans le monde en 2020
(Atlas of MS 3rd edition, september 2020)
Selon les donées 2020 de l’Atlas of MS, La sclérose en plaques est une maladie fréquente dans les pays
industrialisés, et a une prévalence hétérogne dans le monde: élevée en Amérique du nord ( 200/100000
habitants) et en Europe (100/ 100000 habitants), faible en Asie de l’Est et en Afrique (0-25/ 100000
habitants). L’Algérie est classée en prévalence intermédiaire (26-50/100000 habitants)
La prévalence médiane globale de la SEP a augmenté de 33/100000 habitants en 2013 à 44/100000
habitants en 2020, avec un total de 2,8 millions de cas en 2020. Un gradient de prévalence nord-sud a
été décrit pour la distribution de la maladie ( prévalence élevée dans le nord, basse dans le sud).
La SEP est la premire source d’handicap non traumatique chez l’adulte jeune, avec une moyenne d’âge
de 32 ans, tout en sachant qu’il existe des formes pédiatriques et gériatriques. Il existe en autre une
prédominance féminine globale estimée à 3Femmes/1Homme.

3. ÉTIOPATHOGENIE
3.1 ETIOLOGIES
La SEP est une maladie multifactorielle, dont l’origine précise n’est pas encore élucidée. Elle semble
associée à des facteurs géographiques, génétiques et environnementaux. L’association de plusieurs
facteurs est susceptible de déclencher la maladie.
3.1.1. SUSCEPTIBILITE GENETIQUE
La SEP est une maladie chronique non héréditaire de l’adulte jeune, mais il existe une certaine
susceptibilité génétique. Il existe notamment des formes familiales qui restent cependant rares (environ
10%).
Le risque de développer une SEP est deux fois plus important chez les femmes que chez les hommes.
Cependant le pronostic est meilleur chez les femmes. Les études ont montré qu’aucun gne significatif
n’a été décrit sur le chromosome X.
Chez les jumeaux homozygotes, le taux de concordance maximale est de 25%. La prévalence est de
2,75% si l’un des parents est atteint, 4% si la fratrie est touchée (cette incidence est de 0,1% en
population générale).
Une association entre la SEP et des anomalies de la région HLA localisée sur le chromosome 6 est
connue depuis plus de 30 ans.
3.1.2. FACTEURS ENVIRONNEMENTAUX
FACTEURS INFECTIEUX
A ce jour, le virus pour lequel les arguments les plus solides ont été démontrés est le virus de la
mononucléose infectieuse: « Epstein-Barr Virus » (EBV). L’infection primaire par EBV est souvent
asymptomatique à l’âge précoce et se manifeste par une mononucléose infectieuse pendant
l’adolescence ou l’âge adulte et s’accompagne
Le virus EBV présente des similitudes avec la protéine basique de la myéline. Cela semble induire une
confusion du systme immunitaire, qui s’est développé pour neutraliser le virus Epstein-Barr, mais qui
par similitude attaquerait la myéline.
ROLE DE LA VITAMINE D
La vitamine D est bien connue pour son rle dans le métabolisme osseux mais elle possde aussi
d’importantes propriétés de modulation de la réponse immunitaire. La prévalence de la SEP est plus
élevée dans les régions de pauvre ensoleillement. Cette théorie liant le défaut d’exposition solaire et la
SEP est vraie pour d’autres maladies auto-immunes notamment le lupus. Le taux plasmatique de la
vitamine D est effondré chez les patients atteints de SEP et est corrélé au handicap de faon significative.
LE TABAC
Il existe une augmentation significative du risque de survenue de cette pathologie chez les fumeurs. Ce
risque est plus important chez les gros fumeurs que chez les fumeurs classiques. Le tabagisme passif

peut favoriser le déclenchement d’une SEP surtout chez les jeunes enfants subissant le tabagisme des
parents.
LE STRESS
Le stress est un facteur incriminé aussi bien dans le déclenchement ainsi que dans les poussées de la
SEP. L’évaluation du stress vécu par les patients atteints de SEP a été rapportée par plusieurs auteurs et
son implication comme facteur aggravant l’évolution est controversée.
SURPOIDS ET OBSIT
L’obésité crée l’environnement optimal pour les maladies auto-immunes. L’inflammation interne
développée chez les personnes obses est susceptible d’aggraver la progression de la pathologie. Des
expériences menées sur des souris atteintes de SEP ont montré que leur paralysie s’est améliorée aprs
un régime méditerranéen. (25)
VACCINATION
Aucune vaccination n’est contre indiquée. Les grandes études récentes sont rassurantes concernant la
vaccination notamment contre l’hépatite B, une vaccination pour laquelle une polémique a été déclenché
il ya quelques années suite à la description d’un cas découvert lors d’une compagne vaccinale en France.
3.2. PHYSIOPATHOLOGIE
La physiopathologie de la SEP reste encore incompltement connue. La SEP est caractérisée par une
réaction inflammatoire dirigée contre une cible antigénique spécifique: la myéline du SNC et les
oligodendrocytes.
L’inflammation est le mécanisme primitif intervenant dans la sclérose en plaques, la dégénérescence
axonale et neuronale observée est probablement secondaire à ce processus inflammatoire, avec
activation des lymphocytes en périphérie suivie de leur transmigration à travers la barrire hémato-
encéphalique. Une réaction immunitaire est développée dans le SNC impliquant les lymphocytes T,
lymphocytes B, avec production de cytokines et d’anticorps.
Il existe deux grandes hypothses concernant l’activation des cellules auto-réactives:
La premire hypothse est celle du mimétisme moléculaire « cross-réactivité » entre l’agent infectieux
et des cellules du soi. Les cellules immunitaires activées face à l’infection seront alors capables de
reconnaitre les antignes du soi et de s’y attaquer. Les virus probablement responsables sont l’EBV et
l’HHV6. L’autre hypothse est celle des doubles TCR (T Cell Receptor). Selon cette dernire, un virus
activerait des cellules immunitaires possédant 2 TCR: un dirigé contre le virus et l’autre contre un
antigne du soi.
Lors des poussées de SEP, la BHE est fragilisée et partiellement détruite.
La migration des LcT à travers la BHE implique l’interraction des molécules d’adhésion présentes à la
surface des lymphocytes et des cellules de l’endothélium vasculaire. Ce lien est renforcé par l’interaction
des molécules d’adhérence intercellulaire (ICAM-1) et vasculaire (VCAM-1) avec les Ags associés aux
LcT (LFA-1 et VLA-4) dont l’expression à la surface des cellules activées est augmentée.
Les LcT peuvent ainsi traverser l’endothélium et atteindre le parenchyme cérébral sous l’action de
chimiokines. Une fois arrivées dans le SNC, les cellules sont de nouveau réactivées. Cette réactivation
s’effectue suite à la présentation des auto-antignes aux LcT qui librent des composants induisant la
cascade inflammatoire. Les LcB, en dehors de la production d’anticorps via la différenciation en
plasmocytes, les LcB sont responsables de la présentation antigénique aux LcT, induisent une
 6
7
8
9
10
11
12
13
14
15
6
7
8
9
10
11
12
13
14
15
1
/
15
100%