Chapitre I : LES GLYCOPEPTIDES I/ STRUCTURE ET

3
Chapitre I : LES GLYCOPEPTIDES
I/ STRUCTURE ET CLASSIFICATION
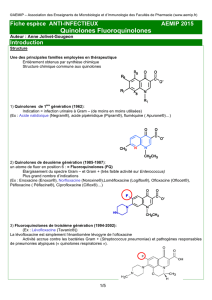
Les glycopeptides sont de volumineuses molécules de haut poids moléculaire
(1450 daltons pour la vancomycine et 1890 daltons pour la teicoplanine). Ce sont
des peptides macromoléculaires tricycliques contenant une chaîne heptapeptidique
linéaire (figure 1) (6).
I-1 LA VANCOMYCINE
La vancomycine (C
66
H
75
Cl
2
N
9
O
24
) est un glycopeptide tricyclique dichloré
possédant une chaîne heptapeptidique comportant cinq cycles aromatiques (partie
active de la molécule), à laquelle est fixé un disaccharide composé de glucose et de
vancosamine.
I-2 LA TEICOPLANINE
La teicoplanine est un complexe de poids moléculaire. Elle est formée de six
composants principaux dont cinq majeurs désignés T
A3
– 1 à T
A2
–5
(Teichomycine
A3
) et un constituant dit « mineur » T
A3
(Teichomycine A
3
). Tous
sont des heptapeptides comportant sept cycles aromatiques.

4
II/ PROPRIETES PHYSICO-CHIMIQUES
II-1 LA VANCOMYCINE
La vancomycine est commercialisée sous forme de chlorhydrate de
vancomycine , lyophilisat pour usage parental intraveineux très soluble dans l’eau
(vancocine® , vancomycine Dakota pharm® , vancomycine Lederle®).
La vancomycine est hydrosoluble si le pH est inférieur à 4 (32). Cette molécule est
irritante pour les veines et est inutilisable par voie intramusculaire.
II-2 LA TEICOPLANINE
Les T
A2
portent plus de trois oses et une chaîne latérale d’acide gras
différents pour chacun des cinq composants. Ceci leur confère une grande
lipophilie, permettant une meilleure diffusion tissulaire d’où une demi-vie longue.
La T
A3
ne compte que deux oses et ne présente pas de chaîne d’acide gras ; il s’agit
du composé le plus polaire.
La teicoplanine est un acide faible, soluble dans l’eau, avec une bonne
tolérance intraveineuse ou intramusculaire.
III/ PROPRIETES PHARMACOLOGIQUES
III-1 LA VANCOMYCINE
Chez l’adulte aux fonctions rénales normales, la dose habituelle est de deux
grammes (2g) (30mg/kg) par jour en administration intraveineuse (à raison de 2 à 4
injections). Le produit est injecté en perfusion lente d’au moins 60 minutes.

5
L’administration rapide ayant des effets secondaires (intolérance locale veineuse :
syndrome de « flush », risque d’arrêt cardiaque lors d’injection intraveineuse.
La posologie doit être réduite chez les insuffisants rénaux, notamment en
raison de l’important allongement de la demi-vie. En pédiatrie, la posologie de
vancomycine est fonction de l’âge ; elle est de 15mg/kg toutes les 12 heures chez le
nouveau-né et de 10mg/kg toutes les 6 heures.
Un taux d’environ 55% de la dose de vancomycine administrée serait fixé
aux protéines sériques. Administrée par voie orale, la vancomycine est faiblement
absorbée à partir du tube digestif et les taux sériques sont très faibles ; par contre,
l’antibiotique atteint des taux élevés dans les selles, ce qui peut constituer une voie
efficace de traitement de la colite pseudomembraneuse due à Clostridium difficile.
Administrée par voie intraveineuse, la demi-vie d’élimination est de 4 à 8 heures
chez le sujet aux fonctions rénales normales. En cas d’insuffisance rénale, elle peut
atteindre 12 jours. Administrée par voie intrapéritonéale, la vancomycine est
rapidement absorbée et passe dans la circulation générale (6).
III-2 LA TEICOPLANINE
La teicoplanine est administrée à la dose de 6 ou 7mg/kg, par voie
intramusculaire ou intraveineuse ; elle est cependant utilisée à la dose de 100mg
deux fois par jour pendant 10 jours dans le traitement de colites
pseudomembraneuses et de diarrhées associées à Clostridium difficile.
Environ 90% du produit est lié aux protéines plasmatiques. Sa demi-vie
d’élimination varie de 40 à 70 heures , ce qui implique l’administration d’une seule
dose toutes les 24 heures ( un avantage sur la vancomycine).

6
Chez les patients hémodialysés, la teicoplanine est administrée une fois par
semaine, avec maintien de taux sériques efficaces. On peut l’administrer par voie
intraveineuse ou intramusculaire.
Environ 95% du produit est éliminé par voie rénale. Il peut être prescrit chez
un patient allergique à la vancomycine (6).
IV/ MECANISMES D’ACTION ET SPECTRE ANTIBACTERIEN
IV-1 MECANISMES D’ACTION
Chez les bactéries à Gram positif, les glycopeptides diffusent au sein de la
paroi bactérienne qui est constituée à 90% de peptidoglycane, et se fixent à leur
substrat : les disaccharides-pentapeptides. Ces disaccharides-pentapeptides sont
synthétisés au sein du cytoplasme de la bactérie. Ils franchissent ensuite la
membrane cytoplasmique pour rejoindre la paroi bactérienne où leur
polymérisation permet la synthèse du peptidoglycane.
Le pentapeptide se termine par une séquence D-alanyl- D-alanyl (D-ala-D-
ala) qui est reconnu par les glycopeptides. Les molécules de glycopeptide ont une
forme tridimensionnelle leur permettant de recouvrir le D-ala-D-ala terminal du
pentapeptide qui n’est plus alors accessible aux enzymes assurant la polymérisation
du peptidoglycane. Cette fixation a pour première conséquence d’inhiber l’action
des carboxypeptidases et des transpeptidases qui ne peuvent plus exciser la D-
alanine terminale du disaccharide-pentapeptide et assurer la liaison du D-analyl
subterminal au résidu du peptidique d’un disaccharide-pentapeptide déjà
polymérisé au sein du peptidoglycane.
De plus, compte tenu de leur masse moléculaire particulièrement élevée, les
glycopeptides empêchent le rapprochement du disaccharide-pentapeptide et de la

7
partie terminale du peptidoglycane qui permettent de créer une liaison entre les
fractions glucidiques des disaccharide-pentapeptides.
Les glycopeptides provoquent ainsi un arrêt de la synthèse du peptidoglycane
et de la croissance bactérienne. Cette activité est bactéricide et lente à l’inverse des
bêta-lactamines. Les glycopeptides sont inactifs sur les bactéries à Gram négatif
dont la paroi est pauvre en peptidoglycane.
IV-2 SPECTRE ANTIBACTERIEN
La plupart des bactéries à Gram positif sont sensibles aux glycopeptides avec
des concentrations minimales inhibitrices (CMI) généralement ou égales à 4µg/ml.
Pratiquement toutes les souches de Staphylococcus aureus restent sensibles à la
vancomycine, qu’elles soient sensibles ou résistantes à la méticilline. Toutes les
souches de pneumocoques testées, incluant des souches pénicillo-résistantes, les
streptocoques des groupes A, B, C et G ainsi que la grande majorité des souches de
Streptococcus viridans et Streptococcus bovis sont sensibles. En France, la majorité
des entérocoques (Enterococcus fœcalis et Enterococcus fœcium) reste sensible aux
glycopeptides. La vancomycine est active contre les diphtéroïdes dont
Corynebacterium jeikeium, Listeria monocytogenes, Borrelia burgdorferi. Seule la
moitié des souches d’Actinomyces est sensible. Nocardia asteroides, les
lactobacilles, les rickettsies, les chlamydiae, les tréponémes, les mycobactéries, les
leptospires, les mycoplasmes, et tous les bacilles à Gram négatif sont résistants.
Le spectre de la teicoplanine est superposable à celui de la vancomycine avec
une activité supérieure sur les entérocoques mais inférieure sur les staphylocoques
à coagulase négative.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
1
/
48
100%