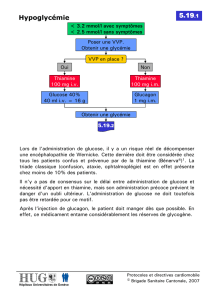
Lire l'article complet

164
Métabolismes Hormones Diabètes et Nutrition (IX), n° 5, septembre/octobre 2005
Urgences
en endocrinologie, diabétologie
et maladies métaboliques
en endocrinologie, diabétologie
et maladies métaboliques
Hypoglycémies chez l’enfant
Hypoglycemia in children
G. Touati*
Définition
L’hypoglycémie se définit par une
glycémie inférieure à 2,8 mmol/l
chez l’enfant et à 2,2 mmol/l chez le
nouveau-né à terme.
L’hypoglycémie est une urgence en
raison du risque de séquelles neuro-
logiques des formes profondes et/ou
prolongées.
Signes cliniques
Les principales manifestations cli-
niques des hypoglycémies sont :
Chez l’enfant
✓
faim impérieuse, douleurs abdo-
minales, nausées, vomissements ;
✓
pâleur, sueurs ;
✓
troubles du comportement, irrita-
bilité, état confusionnel ;
✓
céphalées, troubles visuels ;
✓
regard vide, somnolence, apathie,
coma ;
✓
des convulsions sont très souvent
révélatrices.
Tous ces signes sont non spécifiques,
d’où la nécessité de pratiquer une
glycémie devant tout symptôme
anormal chez l’enfant.
Chez le nouveau-né
✓
pâleur, flush ;
✓
hypothermie ;
✓
cri anormal, irritabilité ;
✓
difficultés d’alimentation, vomis-
sements ;
✓
polypnée, pauses respiratoires ;
✓
tachycardie ;
✓
trémulations, mouvements anor-
maux, hypotonie, somnolence ;
✓
un malaise grave ou une convulsion
sont souvent révélateurs.
Des hypoglycémies, même profondes,
peuvent rester longtemps asympto-
matiques à cet âge, d’où la nécessité
de les dépister par une surveillance
glycémique systématique, en parti-
culier chez le nouveau-né à risque.
Diagnostic
Les signes cliniques étant peu
spécifiques, le diagnostic ne peut
être affirmé que par la mesure de la
glycémie au moment des symptômes
anormaux. Les méthodes de dosage
sur sang capillaire avec un lecteur
de glycémie sont très utiles pour un
diagnostic rapide d’orientation au lit
du malade. Cependant, le diagnostic
doit toujours être confirmé par une
glycémie mesurée au laboratoire par
une méthode de référence.
Éléments cliniques
nécessaires au diagnostic
étiologique
Certains éléments simples permettent
souvent d’orienter rapidement le
diagnostic :
✓L’âge de début,
variable selon les
différentes pathologies.
✓L’ horaire de survenue par rapport
aux repas
est essentiel à connaître :
les hypoglycémies de jeûne évo-
quent des anomalies de la néogluco-
genèse et de la glycogénolyse, des
troubles de l’oxydation des acides
gras, mais aussi la plupart des hypo-
glycémies par déficit hormonal. La
durée de jeûne est un élément clé du
diagnostic des différents déficits
enzymatiques. Les hypoglycémies
postprandiales s’observent au cours
des intolérances au fructose, au
galactose ou au glycérol, mais aussi
dans les hyperinsulinismes leucine-
sensibles et les hyperinsulinismes
poststimulatifs. Les hypoglycémies
sans horaire, anarchiques, sont très
évocatrices des hyperinsulinismes.
Le déficit en hormone de croissance
(GH) survenant essentiellement chez
le petit enfant peut parfois présenter
aussi ce caractère anarchique.
✓L’existence d’une hépatomégalie
est un élément clé du diagnostic.
Une hépatomégalie franche évoque
a priori une anomalie enzymatique
héréditaire.
✓La courbe de croissance
est d’inté-
rêt. Une croissance accélérée fait
suspecter un hyperinsulinisme. Une
cassure de la courbe de croissance
fait évoquer un déficit en GH.
Examens biologiques
nécessaires au diagnostic
étiologique
Les prélèvements doivent être effec-
tués
au moment de l’hypoglycémie
et
permettent alors, le plus souvent,
d’affirmer le diagnostic étiologique.
Le bilan initial comprend :
✓dans le sang :
bilan hépatique,
ionogramme, équilibre acide-base,
lactate, et recueil de 2 ml de plasma
qui permettra l’ensemble des dosages
* Praticien hospitalier, hôpital Necker enfants-
malades, Paris.

Métabolismes Hormones Diabètes et Nutrition (IX), n° 5, septembre/octobre 2005
165
Deuxième partie : urgences métaboliques
hormonaux : insuline, C-peptide,
GH et cortisol, ainsi que l’étude du
profil des acyl-carnitines ;
✓dans les urines :
le recueil de la
première miction qui suit l’hypo-
glycémie permettra d’effectuer une
recherche de cétonurie avec une
bandelette urinaire spécifique et
une chromatographie des acides
organiques.
Principales causes
des hypoglycémies
chez l’enfant
Les hyperinsulinismes primitifs
persistants
se traduisent par des
hypoglycémies sévères, répétées,
d’horaire anarchique, sans cétose et
sans hyperlactacidémie. Elles sont
difficiles à corriger en raison de
besoins importants en glucose. La
réponse au glucagon (i.m. ou i.v.) est
un élément très utile du diagnostic et
de la thérapeutique. L’élévation de
l’insulinémie (> 5 mU/l) au moment
d’une hypoglycémie affirme l’hyper-
insulinisme, mais elle peut être diffi-
cile à mettre en évidence et nécessi-
ter des dosages répétés.
Les hyperinsulinismes transitoires du
nouveau-né
s’observent souvent dans
des situations favorisantes : nouveau-
né de mère diabétique, macrosomie,
souffrance fœtale.
Les insuffisances hypophysaires
à
l’origine d’hypoglycémies survien-
nent surtout dans les deux premières
années de vie. L’existence d’une
cassure de la courbe de croissance,
l’association à un micropénis ou à
une ectopie testiculaire bilatérale
chez le garçon, ou l’association à
une anomalie de la ligne médiane,
peuvent orienter le diagnostic. Le
déficit en GH peut être isolé ou
associé à un déficit en ACTH. Les
hypoglycémies sont très fréquentes
dans le syndrome de Laron lié à une
résistance à l’action de la GH.
L’insuffisance surrénale primaire
peut associer à l’hypoglycémie une
mélanodermie ou une hyponatrémie,
ce qui oriente le diagnostic. Il s’agit
d’hypoglycémies de jeûne ou surve-
nant à l’occasion d’un stress.
Autres déficits endocriniens :
l’hypo-
glycémie est un signe d’hypothy-
roïdie profonde. Le déficit en gluca-
gon est exceptionnel.
Le diagnostic de glycogénoses
se
discute surtout dans les deux pre-
mières années de vie. L’hypogly-
cémie survient au jeûne et s’accom-
pagne de la constitution rapide
d’une hépatomégalie. À l’hypogly-
cémie s’associe, en règle générale,
une hyperlactacidémie. Les plus fré-
quentes des glycogénoses sont le
déficit en glucose-6-phosphatase
(glycogénose type I), en enzyme
débranchante ou amylo-1,6-glucosi-
dase (type III), en phosphorylase
hépatique (type VI), en phosphory-
lase-b-kinase (type IX) et en glyco-
gène-synthase.
Dans les déficits héréditaires de la
néoglucogenèse
(en fructose-1,6-
diphosphatase et en phosphoénol-
pyruvate kinase [PEPCK]), l’hypo-
glycémie survient uniquement à
jeun et s’accompagne d’une hyper-
lactacidémie.
Au cours des déficits de l’oxydation
mitochondriale des acides gras et
des déficits de la cétogenèse,
l’hypo-
glycémie peut survenir en période
néonatale, souvent en contexte de
défaillance polyviscérale. Chez
l’enfant plus grand, l’hypoglycémie
survient après un jeûne prolongé, ou
lors d’une infection. Elle est carac-
térisée par une hypocétonémie asso-
ciée.
L’intolérance au fructose
est une cause
d’hypoglycémie postprandiale.
D’autres maladies métaboliques
peu-
vent être responsables d’hypogly-
cémies. Il s’agit de la galactosémie et
de certaines maladies du métabo-
lisme des acides aminés : leucinose,
tyrosinose type I, acidémie propio-
nique, méthylmalonique. L’hypogly-
cémie n’est pas isolée et accompagne
les autres signes de ces maladies.
Quelques rares cas de déficits de la
chaîne respiratoire mitochondriale
ont été révélés par une hypoglycémie
avec hyperlactacidémie.
L’hypoglycémie récurrente avec
cétose
se manifeste par des hypo-
glycémies débutant le plus souvent
entre 1 et 5 ans. L’hypoglycémie
survient après un jeûne long (12 à
18 heures) et est favorisée par les
infections intercurrentes et le cata-
bolisme.
Les insuffisances hépatiques
ne sont
responsables d’hypoglycémie sympto-
matique qu’à un stade d’insuffi-
sance hépatique préterminale.
Les hypoglycémies induites,
ou
syndrome de Munchhausen par
procuration, doivent être évoquées
devant des hypoglycémies répétées
inexpliquées.
Les autres causes rares d’hypogly-
cémie
incluent l’insuffisance rénale
sévère, les cardiopathies cyano-
gènes, les intoxications alcooliques
aiguës, les intoxications médica-
menteuses (hypoglycémiants oraux,
salicylés, quinine i.v., antipaludéens
type chloroquine, ß-bloquants, etc.),
les hypoglycémies auto-immunes
par apparition d’anticorps anti-insu-
line ou d’anticorps antirécepteurs de
l’insuline.
Traitement
des hypoglycémies
L’hypoglycémie est une
urgence
thérapeutique.
Toute hypoglycémie,
même asymptomatique, nécessite
un traitement rigoureux.
Chez le grand enfant conscient, la
voie orale avec des apports combi-
nant des glucides d’absorption
rapide (sucre et boissons sucrées) et
des glucides d’absorption lente
(pain et féculents), permet le plus
souvent de maintenir une glycémie
normale.
La seconde mesure thérapeutique
est l’injection intraveineuse rapide
de 0,5 à 1 g/kg de glucose sous
forme de glucosé à 30 %. En cas de
coma, et même après le réveil du
malade, il est prudent de maintenir
un apport glucidique continu oral ou
i.v. sur une base de 6-8 mg/kg/mn

Métabolismes Hormones Diabètes et Nutrition (IX), n° 5, septembre/octobre 2005
166
Urgences
en endocrinologie, diabétologie
et maladies métaboliques
en endocrinologie, diabétologie
et maladies métaboliques
de glucose pendant 12 à 24 heures.
Selon l’état clinique de l’enfant et la
cause de l’hypoglycémie, le relais
sera plus ou moins rapidement pris
par voie orale sous surveillance des
glycémies capillaires.
Chez le nouveau-né, la normalisa-
tion de la glycémie doit être régu-
lièrement vérifiée en raison de la
fréquence des hypoglycémies
asymptomatiques à cet âge.
En cas d’hypoglycémies répétées,
une injection de glucagon (1 mg)
intramusculaire constituera un
double test thérapeutique et dia-
gnostique. L’efficacité est rapide
(< 10 minutes) en cas d’hyperinsu-
linisme, et l’administration continue
de glucagon par voie veineuse est
alors un traitement d’urgence très
utile, notamment dans les formes
sévères du nouveau-né.
Si une insuffisance surrénale est
suspectée, la mise en route d’un
traitement substitutif en urgence
peut être justifiée dès que les prélè-
vements sanguins sont faits.
Prévention
de l’hypoglycémie
Elle doit être prévenue et dépistée
dans les situations à risque chez le
nouveau-né :
✓
prématurité ;
✓
dysmaturité ;
✓
macrosomie ;
✓
enfant de mère diabétique ;
✓
souffrance fœtale ;
✓
polyglobulie.
Toutes situations qui imposent une
surveillance systématique des gly-
cémies.
Références
●
De Lonlay P, Castelnau P, Martin D et al. Hypo-
glycémies du jeune enfant : orientation diagnos-
tique. À propos d’une série de 240 cas. Journées
parisiennes de pédiatrie 1998 ; Flammarion
Médecines-Sciences, pp 261-70.
●
Touati G, Ogier de Baulny H. Anomalies héré-
ditaires du métabolisme des glucides. Encycl Med
Chir (Elsevier, Paris), Endocrinologie-Nutrition,
10-364-A-10, 1998, 6 p.
À venir
dans les prochains numéeos
Urgences
en endocrinologie, diabétologie
et maladies métaboliques
Urgences
en endocrinologie, diabétologie
et maladies métaboliques
Troisième partie :
“Hypophyse et surrénales”
À VENIR
dans le prochain numéro
1
/
3
100%