Myoline

Éditorial
Myoline
Bulletin d’information médicale sur les maladies neuromusculaires
L’équipe pluridisciplinaire assurant le suivi médical des patients atteints
d’une maladie neuromusculaire peut jouer un rôle pour les aidants familiaux en
les intégrant dans le projet de soins.
Reconnaître les situations de crise permet de mettre en place les ressources
médicosociales nécessaires à l’amélioration de la qualité de vie des couples
aidants-aidés.
niveau d’intensité (modérée à importante). Un tiers
d’entre eux ont une qualité de vie significativement
moins bonne que celle de la population générale. Dix
pour cent de ces aidants expriment une souffrance
anxiodépressive relativement intense car dépassant le
seuil pathologique.
L’amélioration de la prise en charge des personnes
atteintes de MNM a augmenté leur espérance de vie
et beaucoup vivent à domicile. Assurer les soins quoti-
diens demande aux proches un investissement impor-
tant qui n’est pas sans incidence sur leur santé, leur
vie professionnelle et sociale. Cette étude montre l’im-
portance de se soucier de la santé des proches for-
tement impliqués dans les soins quotidiens. L’emploi
de questionnaires validés, autoadministrés apparaît
utile pour identifi er les personnes de l’entourage du
malade présentant des signes de détresse psycho-
logique et une qualité de vie particulièrement altérée.
Toutes les interactions environnementales matérielles
et affectives susceptibles d’améliorer ou de diminuer
la qualité de vie du couple aidant-aidé doivent être
reconnues.
Diverses études ont cherché à préciser « l’aide aux
aidants » : inclure le malade dans un projet de vie
personnalisé et de soins planifi és, accéder facilement
à un coordinateur de soins, contrôler et accompa-
gner les symptômes du malade, maintenir la qualité
de vie des aidants, rendre disponible des soins de
répit, accompagner et assister les familles dans leurs
décisions pour un projet de soins global adapté à la
progression de la maladie, informer les équipes soi-
gnantes, les aidants et les patients sur la maladie et
ses conséquences, encourager la participation active
des malades et de leur famille dans le milieu asso-
ciatif : fonction bénéfi que de soutien social exercée
par la participation active aux activités organisées
par une association de malades (diminution de 50 à
70 % du stress émotionnel, amélioration du bien-être
psychologique).
n
CR
(1) F. Boyer et coll., Annales de
Réadaptation et de Médecine
Physique, 2005.
Le 6 octobre 2005, le
ministre de la Santé et
des Solidarités, Xavier
Bertrand, a signé un
arrêté sur labellisation de
centres de réfrérence pour
une maladie rare ou un
groupe de maladies rares.
Ainsi, les consultations
multidisciplinaires de Lille
(adultes et enfants) ont
été désignées en tant
que centre de référence
pour les maladies
neuromusculaires (MNM).
C’est le quatrième centre
labellisé, le coordonnateur
est le Pr Thevenon,
clinique de rééducation,
de réadaptation et de soins
de suites, hôpital Pierre
Swynghedauw, CHU de
Lille. Les trois premiers
centres de référence
labellisés pour les MNM,
pour mémoire, l’Institut
de Myologie (associé
aux hôpitaux Trousseau,
Rothschild et Tenon,
coordonnateur : Pr Eymard),
l’hôpital Raymond Poincaré
de Garches (en association
avec les équipes de
Necker, de Créteil et de
Hendaye, coordonnateur :
Pr Estournet), le CHU
de Nice et l’hôpital
de la Timone à Marseille
(troisième pôle,
coordonnateurs :
Pr Desnuelle et Pr Poujet)
avaient été labellisés en
novembre 2004. L’AFM
rappelle que les malades
résidant hors des régions
de ces quatre centres
doivent continuer à se
tourner vers le réseau des
consultations spécialisées.
n
EB
Une étude française(1) a évalué, chez les aidants fami-
liaux de malades adultes atteints de maladies neuro-
musculaires (MNM) et vivant au sein de leur domicile
familial, les liens entre la charge de soins, leur qua-
lité de vie et la santé mentale perçues. Ces aidants
assurent des soins auprès de 59 personnes atteintes
de MNM (dystrophie musculaire de Duchenne ou de
Becker, dystrophie myotonique de Steinert, dystro-
phie facio-scapulo-humérale, amyotrophie spinale
infantile, myopathie des ceintures, dystrophie mus-
culaire congénitale et atteinte myogène ; âge moyen :
32,1 ans, +/– 15,2). Parmi les 59 aidants interrogés
(81% de femmes), on compte 35 parents (31 mères
et 4 pères), 20 conjoints, une sœur, une grand-mère
et 2 amis (âge moyen : 50 ans, +/– 11). Les aidants
ont rempli le questionnaire du « fardeau de Zarit »
(22 items) évaluant la charge de travail matérielle et
affective ressentie par l’aidant principal (scores allant
d’une absence de perception de « fardeau », à légère,
modérée, importante). Le fonctionnement mental, le
bien-être psychologique, la symptomatologie anxieuse
et dépressive ont été mesurés en autoévaluation, res-
pectivement par deux questionnaires de qualité de vie
liée à la santé (SF-36 et GHQ-12) et le Hospital Anxiety
and Depression Scale (HADS).
Un peu plus de la moitié de ces aidants expriment
une charge de soins perçue de troisième à quatrième
n° 81
Novembre/Décembre 2005
mieux cerner
les difficultés des aidants familiaux
1 Prise en charge : mieux cerner les diffi cultés
des aidants familiaux
2 Fin de vie : accompagner l’enfant et sa famille
2 DMD : intrication cardiorespiratoire
2-3 Compte Rendu : IIIe Journées de la Société
Française de Myologie
3 Le mycophénolate mofétil dans la myasthénie :
Un risque accru de lymphome
4 Flash Sciences
Parution
Annonces
Prise
en charge :

2
Fin de vie :
accompagner l’enfant et sa famille
La confrontation à la fin de vie
d’un enfant fait prendre
conscience aux soignants
de toute l’importance de relier le
professionnel et l’humain.
Si les émotions sont totalement
niées, la communication
avec l’enfant, sa famille ou entre
soignants est tronquée, car
l’empathie est insuffisante.
Un travail a été mené au sein d’une équipe
pluridisplinaire de réanimation pédiatrique(1)
afi n de réfl échir à la façon d’accompagner les
enfants dont le pronostic vital est engagé et
leur famille. La spécifi cité des maladies neuro-
musculaires amène à être régulièrement con-
fronté au décès d’enfants. Il en découle une
grande souffrance pour les familles mais aussi
pour le personnel soignant.
Annoncer et expliquer le pronostic est primor-
dial notamment pour adapter le comportement
et l’écoute de l’équipe vis-à-vis de l’enfant et
de sa famille. Le médecin qui effectue cette
démarche auprès des parents veille, pen-
dant ce moment, à être entièrement dispo-
nible et à prendre le temps de les écouter et
de les informer. En dialoguant avec la famille,
le médecin rassure et surtout s’engage sur la
poursuite de soins visant à ce que leur enfant
souffre le moins possible physiquement et
psychologiquement. Il est important que
chaque soignant soit en accord avec l’équipe
et avec lui-même. Chaque situation étant sin-
gulière, chaque enfant et chaque famille uni-
ques, à chaque fois un travail psychologique
est nécessaire, sur le plan personnel et profes-
sionnel, pour accepter la situation présente.
Dès l’annonce du pronostic létal jusqu’aux
derniers instants et même au-delà, la pro-
fondeur et la vérité du lien établi entre les
soignants, l’enfant et ses parents sont essen-
tielles. Il s’agit alors de ne pas fuir et au
contraire de montrer que l’on peut être dans
une relation soignante même si on ne se situe
pas dans la parole de guérison ou à visée
curative.
Trois conditions au moins sont nécessaires
pour que l’accompagnement des enfants en
fi n de vie et de leur famille se déroule le mieux
possible. La première touche l’équilibre psy-
chologique de chaque membre de l’équipe.
Le décès est une séparation irréversible qui
réactive toutes celles, personnelles et profes-
sionnelles, auxquelles chacun est confronté
et dont le travail de deuil est plus ou moins
élaboré.
La deuxième concerne l’équipe qui doit se
réunir pour analyser la situation de chaque
enfant et dire explicitement que le pronostic
vital est engagé. La façon de se situer dans
l’accompagnement est décidée ensemble ce
qui confère une cohérence aux propos tenus
et permet d’éviter les contradictions et les
faux espoirs. Il en découle la possibilité pour
chacun d’assumer son rôle et une plus grande
complémentarité.
La troisième a trait à l’organisation du travail.
Il est souhaitable de l’envisager à deux afi n de
diminuer la solitude, l’angoisse et la culpabilité
de chaque soignant et de favoriser leur soutien
mutuel. La possibilité de parler de ce qui a été
vécu permet de prendre de la distance et amé-
liore la qualité de l’accompagnement.
n
MF
(1) M. Derome, Neuropsychiatrie de l’enfance et de l’adoles-
cence, 2005, 53 : 166-75.
UNE EXPRESSION CONTRÔLÉE
A titre d’exemple, les mécanismes de régu-
lation de l’expression des gènes synaptiques
ont été récemment identifi és. Dans le muscle
adulte, les protéines ayant une fonction
synaptique (comme le récepteur à l’acé-
tylcholine) sont codées par des gènes
dits synaptiques. Ces gènes sont spécifi que-
ment exprimés par quelques noyaux situés
directement sous la terminaison nerveuse
(noyaux sous-neuraux). Ces mêmes gènes
ne sont pas exprimés dans les noyaux situés
plus loin de la jonction neuromusculaire
(noyaux extra-synaptiques). Ce phénomène
s’explique par l’action conjointe de deux
mécanismes régulateurs contrôlés par l’inner-
vation motrice.
D’une part, des facteurs neuraux (agrine
et neurégulines), stimulent localement (dans
les noyaux sous-neuraux) l’expression
des gènes synaptiques. D’autre part, cette
expression est réprimée par l’activité élec-
trique dans les régions extra-synaptiques de
la fi bre musculaire.
Les IIIe Journées Annuelles de
la Société Française de myologie
(SFM) se sont déroulées les
20 et 21 octobre derniers,
à l’Institut de Myologie à Paris.
Cette édition consacrée
à la « jonction neuromusculaire,
biologie et pathologie »
a, notamment, abordé les syndromes
myasthéniques congénitaux et la
myasthénie autoimmune.
IIIe JOURNÉES DE LA SOCIÉTÉ FRANÇAISE DE MYOLOGIE
Compte Rendu
Comprendre et traiter les syndromes myas-
théniques congénitaux (SMC) et la myasthénie
autoimmune (MAI) nécessite de connaître le
fonctionnement de la jonction neuromuscu-
laire (JNM). Grâce à des progrès déterminants
dans les techniques d’imagerie et de micros-
copie, des avancées considérables dans la
compréhension de ces maladies ont été réali-
sées ces dix dernières années.
Cliniqueinfo
DMD :
intrication respiratoire
Les atteintes cardiaques et pulmonaires
sont systématiques chez les patients
atteints de dystrophie musculaire de
Duchenne (DMD). La cardiomyopathie est
en règle diffuse, plus ou moins précoce,
très longtemps asymptomatique quelle
que soit la sévérité de l’atteinte. En effet,
l’atteinte cardiaque, bien que présente
sur les investigations complémentaires,
reste habituellement asymptomatique jus-
qu’à l’adolescence voire à des stades plus
tardifs de la maladie. Elle devient sympto-
matique chez environ 60% des patients et
la moitié d’entre eux rapportent des symp-
tômes au-delà de 18 ans.
Impliquant d’abord le ventricule gauche, la
cardiomyopathie dilatée peut entraîner une
dyspnée et d’autres symptômes d’insuf-
fi sance cardiaque congestive. Ces symp-
tômes peuvent être communs à l’atteinte
respiratoire et confondus avec celle-ci.
Inversement, l’insuffisance respiratoire
chronique de type restrictif peut engen-
drer une hypoxémie et une hypercapnie,
s’aggravant notamment pendant le som-
meil (importance des explorations noc-
turnes). Il peut en résulter une hypertension
artérielle pulmonaire et une insuffisance
ventriculaire droite. De plus, les patients
atteints de DMD risquent de développer
des troubles du rythme (arythmies ventri-
culaires) mettant en jeu le pronostic vital.
Une dysfonction systolique et des trou-
bles du rythme ventriculaires précoces
sont des facteurs prédictifs de mortalité.
Si certaines études suggèrent que la fai-
blesse musculaire respiratoire et périphé-
rique tend à être inversement liée au risque
d’insuffisance cardiaque, d’autres sug-
gèrent que l’insuffisance respiratoire et
l’insuffisance cardiaque gauche tendent
à survenir parallèlement.
Une évaluation régulière et systématique de
l’état cardiaque (ECG, holter, échographie,
scintigraphie) ainsi qu’une vigilance parti-
culière en cas d’intervention chirurgicale,
de symptômes (malaise, palpitations…) et
d’insuffi sance respiratoire non compensée
permettent de mettre en œuvre une prise
en charge adaptée. Au stade asympto-
matique, l’électrocardiogramme montre
différentes anomalies prédictives de la
pathologie cardiaque (tachycardie sinu-
sale, grandes ondes R en V1, profondes
ondes Q dans plusieurs dérivations, aug-
mentation de la dispersion de l’intervalle
QT). L’échocardiographie peut mettre en
évidence une dysfonction systolique dès
la première décade de vie. La scintigra-
phie (mesure de la fraction d’éjection iso-
topique), moins sensible aux déformations
thoraco-vertébrales, apporte des informa-
tions supplémentaires notamment s’il y
a un doute à l’échographie. Chez environ
90% des patients atteints de DMD, il existe
une atteinte cardiaque asymptomatique ou
cliniquement avérée.
n PL, EB
JD. Finder et coll., American Journal of Respiratory &
Critical Care Medecine, 2004, 170(4) : 456-65.
J. Finsterer et coll., Cardiology, 2003, 99 : 1-19.
Psychologie

Novembre/Décembre 2005
Myoline
Bulletin d’information médicale sur les maladies neuromusculaires
n° 81
AFM
BP 59
91002 EVRY Cedex
Bulletin d’abonnement
3
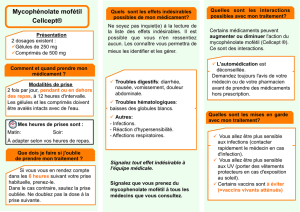
Malgré des effets secondaires modérés à court terme et une
efficacité évidente, le traitement au long cours de la myasthénie par
le mycophénolate mofétil peut être associé à un risque accru de
maladies lymphoprolifératives. En effet après trois ans de traitement,
une patiente, âgée de 83 ans, a développé un lymphome primaire du
système nerveux central. C’est le premier cas rapporté en dehors
d’une transplantation d’organe. L’âge avancé et la durée du traitement
peuvent avoir augmenté le risque.
Le mycophénolate mofétil (cellcept®) est un
agent immunosuppresseur. Il est indiqué en
association à la ciclosporine et aux corticoïdes
pour la prévention des rejets aigus d’organe
chez les patients ayant bénéfi cié d’une allo-
greffe rénale, cardiaque ou hépatique. Il est
aussi utilisé dans le traitement de la myas-
thénie.
Des études ont, en effet, montré que 50 à 70%
des patients (atteints de myasthénie) répon-
dent au mycophénolate mofétil sans présenter
d’effets secondaires graves.
Une équipe américaine rapporte le cas d’une
patiente diabétique, âgée de 83 ans, ayant
développé une myasthénie : diplopie inter-
mittente, ptosis, fatigabilité et faiblesse mus-
culaire des extrémités et dysarthrie. Malgré
un traitement associant pyridostigmine et
prednisone, elle présentait, deux ans plus
tard environ, une perte progressive de la force
musculaire, une augmentation de la fatigabi-
lité, une dyspnée à l’effort et une dysphagie.
Le mycophénolate mofénil a alors été admi-
nistré à la dose de 1 gramme deux fois par
jour. Après six semaines, une amélioration cer-
taine de la force musculaire a été rapportée
par la patiente, entraînant une diminution des
doses de pyridostigmine et de prednisone. La
dyspnée et la dysphagie ont été totalement
résolues. Quant à la dysarthrie, elle était notée
simplement à la fin de la journée. Au cours
des six mois suivants, la dose quotidienne de
prednisone a encore été réduite et la pyridos-
tigmine interrompue. Le seul symptôme per-
sistant était un léger ptosis droit survenant en
fi n de journée.
Pendant deux ans, la réponse à ce traitement
s’est avérée satisfaisante sans effet secon-
daire notable, le nombre des leucocytes res-
tant dans les limites de la normale et celui des
lymphocytes tout à fait acceptable. La sur-
venue d’une lymphocytopénie et d’une anémie
modérée ont justifi é la diminution de la dose
de mycophénolate mofétil à 0,5 gramme deux
fois par jour durant un mois.
Après trois ans de traitement, la patiente,
alors âgée de 88 ans, a présenté des cépha-
lées modérées, une fi èvre légère et une diffi -
culté à trouver les mots. L’IRM a montré une
lésion fronto-pariétale gauche circonscrite par
un œdème et plusieurs lésions satellites. Une
analyse de la lésion principale a révélé un lym-
phome à cellules B. Aucun signe de lymphome
n’a été détecté à l’examen du thorax, de l’ab-
domen, du pelvis, du sang et de la moelle
osseuse. Le diagnostic de lymphome primaire
du système nerveux central lié à l’immunosup-
pression a été établi.
L’administration de mycophénolate mofétil
a été interrompue. Six semaines plus tard, à
l’IRM, l’aspect de la tumeur était nettement
amélioré et le nombre des lymphocytes était
normalisé. Au cours des neuf mois suivants,
l’aphasie et l’hémiparésie droite ont régressé.
Le suivi IRM a montré une résolution pratique-
ment complète du lymphome primaire du sys-
tème nerveux central.
Les effets indésirables, les plus fréquents,
induits par le mycophénolate mofétil sont des
troubles digestifs (notamment diarrhée), une
diminution de la résistance aux infections due
à des troubles hématologiques (leucopénie),
des douleurs, de la fi èvre, des céphalées, une
hypertension artérielle et des oedèmes. Par
ailleurs, les personnes recevant du mycophé-
nolate mofétil sont exposées à un risque accru
de lymphome et de tumeurs malignes notam-
ment cutanées.
S.Vernino et coll., Neurology, 2005, 65 : 639-41
Prescrire...
Le mycophénolate mofétil dans la myasthénie :
un risque accru de lymphome
UN RÉSEAU SMC
Les syndromes myasthéniques congénitaux
(SMC) sont des maladies héréditaires chroni-
ques, rares et invalidantes, débutant souvent
dès l’enfance et pouvant mettre en jeu le pro-
nostic vital.
Ils forment un groupe d’affections hétérogènes
tant sur le plan clinique que génétique, ce qui
explique la diffi culté d’un diagnostic physiopa-
thologique complet.
En 2001, un réseau clinique et de recherche
sur les SMC a été mis en place, réunissant les
services cliniques de neuropédiatrie et de neu-
rologie adulte et des laboratoires de recherche
spécialisés dans l’étude de la jonction neu-
romusculaire. Il a permis de développer pour
la première fois en France l’approche et les
moyens nécessaires à une caractérisation des
SMC reposant sur la clinique, l’électromyogra-
phie, l’étude des jonctions neuromusculaires
sur biopsie musculaire et l’analyse génétique.
Le réseau SMC a ainsi étudié les 100 patients
recensés dans la population française. Un dia-
gnostic moléculaire a pu être donné pour 55
d’entre eux permettant un traitement adapté
et, pour certains, un conseil génétique.
MYASTHÉNIE ?
Plus de 80% des personnes atteintes de
myasthénie autoimmune fabriquent des auto-
anticorps se fi xant sur les récepteurs de l’acé-
tylcholine (RACh). Parmi celles sans anticorps
anti-RACh, 41% ont des anticorps anti-MuSK.
Ces dernières se différencient par la résistance
au traitement par anti-cholinestérasiques.
Enfi n, certains patients atteints de myasthénie
n’ont pas d’anticorps anti-RACh ni d’anticorps
anti-MuSK.
La recherche de ces anticorps est importante,
notamment, dans la myasthénie autoimmune
néonatale, représentant 10% des nouveaux-
nés de mères myasthéniques. La présence
d’anticorps anti-RACh ou anti-MuSK chez un
enfant présentant d’emblée une symptoma-
tologie sévère et chez sa mère permet d’éta-
blir le diagnostic de myasthénie néonatale et
de faire le diagnostic différentiel avec un syn-
drome myasthénique congénital (SMC). En
exemple, le cas d’une patiente âgée de 26
ans présentant une diplopie et un pstosis fl uc-
tuants depuis l’âge de 22 ans qui a accouché,
par césarienne (hydramnios), d’un enfant
hypotonique, avec stridor et troubles respira-
toires nécessitant une intubation. La gravité
des symptômes observés chez l’enfant ainsi
que l’absence d’anticorps anti-RACh chez la
mère et son fi ls, ont orienté vers un SMC. Des
analyses supplémentaires ont, en fait, révélé la
présence d’anticorps anti-MuSK chez l’un et
l’autre, confi rmant le diagnostic de myasthénie
autoimmune.
Des facteurs génétiques (antigènes HLA-B8 et
DR3) sont associés à la forme de myasthénie
touchant les femmes jeunes et comportant
une hyperplasie thymique. Plus récemment, il
a été montré que le gène CHRNA1 codant la
sous-unité alpha de RACh, est associé à une
forme de myasthénie débutant très précocé-
ment. Des travaux ont révélé, chez l’animal,
un effet aggravant des œstrogènes dans la
maladie.
n AR

Reproduction sans but lucratif autorisée en mentionnant l’origine : Myoline, Bulletin d’information médicale sur les maladies neuromusculaires, AFM BP59 91002 Evry Cedex
4
Myoline
AFM - BP59 - 91002 Evry Cedex
Directeur de la publication : Laurence TIENNOT-HERMENT
Directeur de la rédaction : Tuy Nga BRIGNOL
Rédacteur en chef : Edwige BIARD
Maquette : Isabelle PEREIRA
email rédaction : [email protected]
Ont collaboré à ce numéro :
Patrick Léger
Aurélias Reis
Christian Réveillère
Impression : Taag - 01 69 25 40 40
Dépôt légal : Octobre 2005
I.S.S.N. : 1169-5498
LGMD2A :
spectre élargi
A ce jour, 17 formes de dystrophies muscu-
laires des ceintures (LGMD) ont été géné-
tiquement identifiées. Une équipe italienne
a analysé le gène CAPN3 (calpaïne 3) chez
530 sujets atteints de LGMD et 300 sujets
contrôles. Quatre-vingt deux mutations de
CAPN3, dont 45 nouvelles, ont été ainsi iden-
tifi ées chez 141 patients. Les mutations de la
calpaïne 3 ont donc été retrouvées dans plus
d’un tiers des cas de LGMD, mais aussi chez
18,4% des patients atypiques et 12,6% des
sujets avec un taux de CK élevé. La LGMD2A,
classiquement liée à des mutations de CAPN3
apparaît avoir un spectre phénotypique très
varié. Il est aussi montré que dans la popula-
tion étudiée, une personne sur 103 est por-
teuse d’une mutation de CAPN3.
Piluso et coll., J Med Genet, 2005, 42(9) : 686-93
DMED-X :
cœur à suivre
Selon une étude japonaise, une forme de
dystrophie musculaire d’Emery-Dreiffus liée
à l’X associée à une mutation non sens du
gène STA est caractérisée par un tableau cli-
nique atypique : symptômes musculaires très
modérés ou inexistants et complications car-
diaques sévères (bradychardie nécessitant
l’implantation d’un pacemaker, voire fi brillation
auriculaire) pouvant entraîner une mort subite.
Un suivi régulier chez les familles porteuses de
ce type de mutation est préconisé.
Sakata et coll., Circulation, 2005, 111(25) : 3352-8
Atlas des MNM :
un guide pratique
Destiné aux médecins, neurologues et étu-
diants non spécialisés dans les maladies neu-
romusculaires, cet « atlas neuromusculaire »
est censé aider au diagnostic des maladies
neuromusculaires et à tous les niveaux du sys-
tème nerveux périphérique. Il a été rédigé par
des neurologues américains et européens.
Le premier chapitre décrit les outils utilisés
pour établir un diagnostic (interrogatoire, exa-
mens clinique et de laboratoire, électrophy-
siologie, biopsie et génétique). Les chapitres
cliniques commencent par les nerfs craniaux,
suivent les radiculopathies, les mononeuro-
pathies des membres supérieurs/inférieurs
et du tronc ainsi que les polyneuropathies.
Viennent ensuite les maladies de la transmis-
sion neuromusculaire, les pathologies mus-
culaires et myotoniques puis, les maladies du
motoneurone. Le dernier chapitre est une aide
au diagnostic différentiel, il vise à identifi er les
signes et symptômes neuromusculaires asso-
ciés à une pathologie plus générale (diabète,
cancer…). Chaque sujet est abordé selon le
même plan : localisation anatomique, signes
et symptômes, différentes étiologies, dia-
gnostic, diagnostic différentiel, thérapie, pro-
nostic, références bibliographiques.
Les schémas simples et les photos (histologie,
patients…) sont une part essentielle de cet
ouvrage bien structuré, aisément compréhen-
sible et offrant une revue synthétique et claire
des pathologies neuromusculaires.
Feldman, Grisold, Russell, Zifko : « Atlas of neuromuscular di-
seases : a practical guideline”, SpringerWienNewYork, 2005,
474p, prix : 211,86 euros.
ENMG
2006
Les 15e journées francophones d’électroneuro-
myographie se tiendront à Grenoble du 15 au
17 mars prochain. Parmi les thèmes abordés
en conférences, on note les neuropathies,
myopathies, canalopathies et les pathologies
du motoneurone ainsi que les stratégies
d’exploration et les recommandations de
bonne pratique. Des tables rondes traiteront
de l’interprétation des données ENMG et de la
place de celle-ci, en 2006, dans les maladies
neuromusculaires.
Renseignements : BCA, 6 boulevard du général Leclerc, 92115
Clichy Cedex. Tél : 01 41 06 67 70. Fax : 01 41 06 67 79.
E-mail : [email protected] Site Internet : www.b-c-a.fr
Journées
de Neurologie
Les journées de neurologie de langue fran-
çaise auront lieu, à Toulouse au Centre de
Congrès Pierre Baudis, du 12 au 15 avril 2006.
Sclérose en plaques, céphalées-migraines,
pathologie vasculaire, syndromes parkinso-
niens, neurogénétique et actualités thérapeuti-
ques sont les thèmes programmés. Durant ces
3 jours d’enseignement supérieur de la neu-
rologie, des ateliers pratiques sont aussi pro-
posés (inscription préalable payante).
Renseignements : BCA, 6 boulevard du général Leclerc, 92115
Clichy Cedex. Tél : 01 41 06 67 70. Fax : 01 41 06 67 79.
E-mail : [email protected] Site Internet : www.b-c-a.fr
Arthrodèse : la ventilation,
une priorité en postopératoire
Les techniques de rééducation respiratoire (ventilation par
percussions et hyperinsuffl ations) couplées à la ventilation
non invasive par voie nasale facilitent le sevrage de l’intuba-
tion endo-trachéale et la récupération de la capacité vitale
après arthrodèse. Tels sont les résultats d’une étude menée,
par une équipe de l’hôpital Raymond Poincaré (Garches)(1),
chez quatre vingt quatorze patients atteints de maladies
neuromusculaires hospitalisés pour une arthrodèse verté-
brale. Ces enfants, âgés en moyenne de 13 ans et demi lors
de l’intervention, ont été intubés pendant environ 10 jours
en postopératoire. Pendant et après l’intubation, ils ont été
traités par des hyperinsuffl ations et une ventilation par per-
cussions. Une ventilation non invasive par voie nasale a été
mise en place chez 39 % des patients après extubation.
Au total, 70% des patients ont retrouvé leur capacité vitale
préopératoire avant la fin de l’hospitalisation, les autres
l’ayant récupérée dans un délai de deux à six mois après
l’intervention.
Rappelons que l’arthrodèse rachidienne, chez un patient
atteint de maladie neuromusculaire, est généralement suivie
d’une perte de 50% de sa capacité vitale préopératoire. Il
en résulte que, non seulement la durée d’intubation est plus
longue que pour d’autres interventions mais aussi qu’une
prise en charge spécifi que est nécessaire à la récupération
des capacités ventilatoires préopératoires.
(1) M. Hamida et coll., communication libre lors du 9e congrès Méditerranéen de
Ventilation Non Invasive & Actualités en Réanimation (Lyon, 15 et 16 novembre
2005).
PourInfo
BULLETIN D’ABONNEMENT
n
Je m’abonne GRATUITEMENT à Myoline
pour 1 an :
Nom .................................................................
Prénom ............................................................
Profession/Spécialité .........................................
........................................................................
Adresse et code postal .......................................
........................................................................
........................................................................
La loi informatique et liberté du 06/01/78, vous permet d’exercer les droits d’accès,
de rectifi cation et de radiation prévus aux articles 26, 34 et 36 en vous adressant à
l’AFM - BP 59 - 91002 EVRY Cedex.
FlashSciences Parution
Annonces
1
/
4
100%