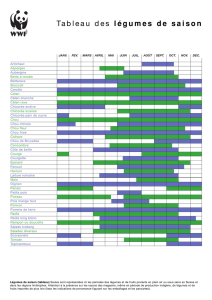
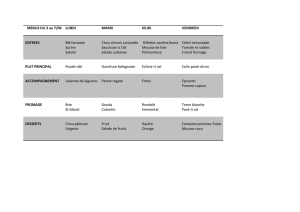
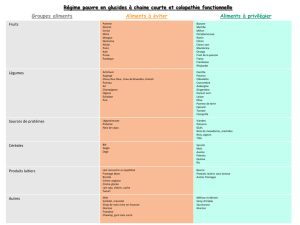
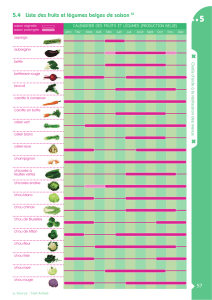
Classification chimique des médicaments

L2 Pharmacie – Chimie thérapeutique N°2
19/03/14 – Pr Dallemagne
Groupe 17 – Chou et Chou
1
Classification chimique des médicaments
Les « drug prototypes » et leur filiation
I. L’Exemple de la Filiation de la Chrysoïdine
Q. Clofénamide
R. Chlorothiazide
S. Furosémide
T. Clopamide
U. Amiloride
V. Carbutamide
W. Tolbutamide
X. Chlorpropamide
Y. Glicazide
Z. Acétohexamide
Z’ . Glibenclamide
II. L’Exemple de la Filiation de l’Adrénaline
A. Synéphrine et Phényléphrine
B. Isoxuprine
C. Isoprénaline
D. Orciprénaline
E. Fénotérol
F. Terbutaline
G. Bambutérol
H. Salbutamol
I. Pirbutérol
J. Salmétérol
K. Dichloro-isoprénaline
L. Pronéthanol
M.Propranolol
N. Nadolol
O. Timodol
P. Alprénolol
Q. Oxprénolol

L2 Pharmacie – Chimie thérapeutique N°2
19/03/14 – Pr Dallemagne
Groupe 17 – Chou et Chou
2
I. Exemple de la filiation de la Chrysoïdine
Q. Clofénamide.
Carzénide : propriétés salidiurétiques.
Les scientifiques de MSD découvrirent que la présence d’un
deuxième groupement sulfonamide dans la structure du carzénide
augmentait la capacité de celui-ci à accroître l’excrétion urinaire des
ions Na+.
Le remplacement du groupement acide carboxylique par un atome
de chlore va dans le même sens.
Introduction en 1958 du clofénamide en tant que puissant
inhibiteur de l’anhydrase carbonique mais davantage
salidiurétique que bicarbonatiurétique.
R. Chlorothiazide

L2 Pharmacie – Chimie thérapeutique N°2
19/03/14 – Pr Dallemagne
Groupe 17 – Chou et Chou
3
Des chercheurs de chez Merck découvrent que l’installation d’un groupement aminé sur
le noyau benzénique du clofénamide diminue l’activité inhibitrice de l’anhydrase
carbonique mais pas l’excrétion des ions Na+.
Synthèse de nombreux analogues avec divers substituants sur le nouveau
groupement amino dont le groupement formyl qui, de façon inattendue subit une
cyclisation intramoléculaire, en dérivé benzothiadiazine appelé chlorothiazide. Ce
n’est plus un inhibiteur de l’anhydrase carbonique.
On le teste quand même et on découvre avec surprise que ce
chlorothiazide est un salidiurétique puissant qui n’augmente pas
l’excrétion des ions bicarbonates.
Le chlorothiazide est introduit en 1957, il devient le chef de file des
diurétiques thiazidiques même s’il n’est plus utilisé en France.
Il a rendu obsolète les diurétiques mercuriels dans le traitement des
œdèmes cardiaques et de l’insuffisance cardiaque congestive. Mais
surtout, il a confirmé l’idée de Beyer qu’un diurétique qui augmente
l’excrétion de NaCl pouvait être d’un grand intérêt dans le traitement
de l’HTA. C’est encore aujourd’hui l’indication principale des
diurétiques thiazidiques.
On est parti de sulfamides d’antibiotiques, qu’on a mis en évidence avec des effets
secondaires indésirables d’ordre diurétique, on a voulu amplifier cet effet, et on a réussi en
modifiant les molécules, en passant de l’effet bicarbonatiurétique à un effet salidiurétique.
En 1958, De Stevens (Ciba) prépare l’hydrochlorothiazide en remplaçant HCO2H utilisé
dans la cyclisation intramoléculaire par HCHO.
L’hydrochlorothiazide est 10 fois plus actif que le chlorothiazide et a une durée d’action
identique. Il est présent dans de nombreuses spécialités.

L2 Pharmacie – Chimie thérapeutique N°2
19/03/14 – Pr Dallemagne
Groupe 17 – Chou et Chou
4
Synthèse de chlorothiazide par Ciba en 1960 :
Il est très puissant : les patients n’absorbent qu’ 1/100 de la dose utilisée avec
l’hydrochlorothiazide (pour réduire les éventuels effets indésirables).
Chlorothiazide
Hydrochlorothiazide CAPTEA ® Cyclopenthiazide CYCLOTHERIAM ®
S. Furosémide
En 1962, les chercheurs de chez Hoechst (laboratoire allemand)
découvrent que l’on pouvait remplacer l’un des groupements acides
(sulfonamides) du clofénamide par un groupement acide
carboxylique pour peu que l’on installe un substituant adéquat en
ortho. C’est la découverte d’un médicament carrefour dans la
famille des diurétiques.

L2 Pharmacie – Chimie thérapeutique N°2
19/03/14 – Pr Dallemagne
Groupe 17 – Chou et Chou
5
Introduction du furosémide qui agit plus rapidement et de façon plus intense que
les autres diurétiques.
Il agit au niveau de l’anse de Henlé = premier diurétique de l’anse commercialisé. Il est
indiqué en cas d’œdème pulmonaire consécutif à une insuffisance ventriculaire
gauche.
Le LASILIX® demeure très prescrit.
T. Clopamide
Le clopamide est introduit par Sandoz en 1962 et présente la
même activité que le chlorothiazide.
La fonction sulfamide est remplacée par une fonction acide
carboxylique dans un groupement carboxamide.
U. Amiloride
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
1
/
21
100%
![Calendrier-fruits-legumes-fevrier[1]](http://s1.studylibfr.com/store/data/004576337_1-b30f140db474af20a4438177a165d730-300x300.png)