Table des matières

Table des matières
Table des matières i
1 LES ALCANES 1
1.1 Nomenclature ..................................... 2
1.2 Isoméries ....................................... 4
1.3 Propriétés physiques ................................. 5
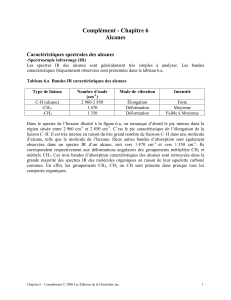
1.4 Propriétés spectrales .................................. 6
1.5 Réactions chimiques des alcanes ........................... 17
1.6 Synthèse des alcanes et des cycloalcanes ....................... 26
1.7 Exemples d’alcanes .................................. 27
Bibliographie 32
i


Chapitre
1LES ALCANES
1.1 Nomenclature................................ 2
1.2 Isoméries .................................. 4
1.3 Propriétésphysiques ............................ 5
1.4 Propriétésspectrales............................. 6
1.5 Réactions chimiques des alcanes . . . . . . . . . . . . . . . . . . . . . . 17
1.6 Synthèse des alcanes et des cycloalcanes . . . . . . . . . . . . . . . . . . 26
1.7 Exemplesd’alcanes............................. 27
Figure 1.1 – Le méthane
L
es hydrocarbures aliphatiques saturés, ou alcanes, ont pour formule générale :
CnH2n+2
. Ils
ne possèdent pas de groupement fonctionnel proprement dit et sont exclusivement constitués
de liaisons simples non polaires
C−C
et
C−H
. On les représente souvent par le symbole
général : R−Hdans lequel Rdésigne un radical alkyle, et on les appelle également paraffines.
Figure 1.2 – Exemples d’alcanes
1

2LES ALCANES
1.1 Nomenclature
La nomenclature des alcanes sert de base à celle de toutes les fonctions en série aliphatique.
Les alcanes sont désignés par le suffixe -ane : méthane (
CH4
), éthane (
C2H6
), propane (
C3H8
),
butane(
C4H10
), pentane (
C5H12
), hexane (
C6H14
), heptane (
C7H16
), octane (
C8H18
)... Ces dénomi-
nations supposent que la chaîne de carbones est linéaire, série dite normale. Les groupes alkyles
correspondants formés par la perte d’un atome d’hydrogène sur un carbone terminal sont appelés
respectivement méthyle (
CH3
ou Me), éthyle (
C2H5
ou Et), n-propyle (
n−C3H7
ou n-Pr), n-butyle
(C4H9ou n-Bu),
Dans les systèmes bicycliques deux atomes de carbone sont communs à deux cycles. Ils sont nommés
en énonçant le préfixe « bicyclo » devant l’hydrure fondamental acyclique qui comporte le nombre
total d’atomes du squelette. Les hétéroatomes sont indiqués, le cas échéant, par la nomenclature
par remplacement en utilisant des termes en « a ». Le nombre d’atomes du squelette de chacune des
trois chaînes acycliques (ponts) reliant les deux atomes communs (tête de pont) est indiqué par des
chiffres arabes énoncés dans l’ordre décroissant, séparés par des points et placés entre crochets.
La numérotation commence par un atome tête de pont suivant la branche la plus longue. Elle se
poursuit le long de la branche la plus courte en passant par le deuxième tête de pont pour terminer
vers le pont principal.

Nomenclature 3
Pour les composés spirocycliques, les anneaux ont en commun un seul atome de carbone. Ils
sont nommés en plaçant « spiro » devant le nom de l’hydrure fondamental acyclique possédant
le même nombre d’atomes du squelette ; les hétéroatomes sont désignés par la nomenclature par
remplacement, c’est-à-dire au moyen de préfixe placés devant le préfixe spiro. Les nombres d’atomes
du squelette liés à l’atome spiranique dans chacun des cycles sont indiqués par des chiffres arabes
séparés par des points, cités dans l’ordre croissant et placés entre crochets. Ce descripteur est placé
entre le préfixe spiro- et le nom de l’hydrure fondamental. La numérotation commence par un atome
cyclique voisin de l’atome spiranique et se poursuit d’abord par le plus petit cycle, puis par l’atome
spiranique et enfin par le second cycle.
Le tableau 1.1 résume les préfixes en «a»utilisés en nomenclature par remplacement, pour quelques
éléments.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
1
/
35
100%