L Avancées dans le cancer du côlon héréditaire :

LE GÈNE DU MOIS
84
La Lettre du Cancérologue - Volume XIV - n° 2 - mars-avril 2005
L
e cancer du côlon héréditaire (syndrome HNPCC,
OMIM #114500) est une pathologie actuellement
bien caractérisée mais qui reste largement sous-diag-
nostiquée. Aux États-Unis, par exemple, on estime qu’un
screening systématique de l’instabilité des microsatellites
chez les 140 000 nouveaux cas annuels de cancer du côlon
permettrait d’identifier 21 000 nouveaux cas (patients atteints
et apparentés) porteurs d’une mutation causale du syndrome
HNPCC (1). L’identification des porteurs d’une mutation
d’un des gènes responsable du syndrome HNPCC (MLH1,
MSH2, MSH6, appelés aussi gènes MMR) est importante
cliniquement, puisque la surveillance par coloscopie de ces
porteurs permet de réduire de façon substantielle la mortalité
due aux cancers colorectaux (2). On estime que le syndrome
HNPCC est responsable d’environ 2 % des cancers colorec-
taux (tableau I).
Plusieurs stratégies diagnostiques ont été proposées récem-
ment, mais l’évaluation de leur sensibilité n’a pas été réali-
sée (3). Cet article décrit les performances de différents cri-
tères cliniques, présente la nouvelle version des critères de
Bethesda et discute l’apport potentiel d’un nouveau marqueur
moléculaire dans la tumeur, une mutation du gène BRAF
(OMIM #164757).
PERFORMANCE DES CRITÈRES DIAGNOSTIQUES
CLASSIQUES
Les critères diagnostiques établis initialement pour permettre
l’identification des gènes responsables du syndrome HNPCC se
révèlent trop restrictifs en pratique courante, avec un manque de
sensibilité important. Les premiers critères établis étaient les cri-
tères dits d’Amsterdam I (tableauII), définis en 1991 (4). Cepen-
dant, les études des familles avec un syndrome de Lynch ont rapi-
dement révélé la présence d’un excès de cancers de l’endomètre,
de l’estomac, de l’intestin grêle, du système hépatobiliaire, des
bassinets rénaux et des uretères, ainsi que des ovaires (5). C’est
pourquoi les critères d’Amsterdam II ont été revus en 1997,
incluant dans leur définition les patients atteints d’un cancer
Avancées dans le cancer du côlon héréditaire :
les critères révisés de Bethesda et le gène BRAF
News in hereditary non polyposis colon cancer:
the revised Bethesda criteria and BRAF
●
C. Monnerat*
* Service cantonal multisite d’oncologie, hôpital de La Chaux-de-Fonds,
Suisse.
Tableau I.
Fréquence des cas HNPCC.
Auteur Lieu Population n Stratégie diagnostique I (%)
Salovaara (15) Helsinki Série de CCR non sélectionnés 1 044 MSI →MLH1/MSH2 2,7
Hemminki (16) Suède Registre familial de CCR < 62 ans 63 698 CCR des parents →CCR des enfants 1-2,5
Katballe (17) Danemark Série de CCR non sélectionnés 3 722 Amsterdam I et II et/ou MLH1/MSH2 1,7
Aaltonen (18) Finlande Série de CCR non sélectionnés 1 328 MSI →MLH1/MSH2 2
CCR : cancer colorectal ; I : incidence des cas HNPCC ; n : nombre de cas dans l’étude.
Tableau II.
Critères d’Amsterdam I.
✓ Au moins 3 apparentés avec un cancer colorectal
✓ Au moins un des cas est un apparenté au 1er degré des deux autres
✓ Au moins deux générations sont affectées
✓ Au moins un des cas a été diagnostiqué avant l’âge de 50 ans
✓ Le diagnostic de polypose adénomateuse familiale (FAP) doit être exclu
✓ Les diagnostics doivent être confirmés par un examen histologique
Adapté de Vasen et al., 1991 (4).

colorectal, de l’endomètre, de l’intestin grêle et des voies uri-
naires (tableau III) (6). Une méta-analyse récente a évalué la
performance des critères d’Amsterdam I et II (tableau IV) (7).
La spécificité des critères d’Amsterdam I semble meilleure que
celle des critères d’Amsterdam II, ce qui était attendu, puisque
l’élargissement des critères conduit à analyser davantage de
familles. Cette spécificité sera encore plus grande avec l’amé-
lioration des techniques d’analyse moléculaire, notamment par
la détection des anomalies de grande taille, comme les réarran-
gements. Une étude américaine récente a identifié une mutation
des gènes MLH1, MSH2 ou MSH6 chez 92 % des familles répon-
dant aux critères d’Amsterdam I (8). Dans cette série, la moitié
des mutations du gène MSH2 était des anomalies de grande taille.
Étonnamment, la sensibilité des études rapportées dans cette
méta-analyse était également bonne, oscillant entre 64 % et 83 %.
Il s’agit ici d’un biais de sélection, puisque toutes ces familles
ont été collectées par des centres d’oncogénétique. Un travail
récent analysant les mutations de MLH1 et MSH2 dans deux
populations de patients présentant un cancer du côlon avant 36 ans
a retrouvé des mutations chez 35 % des patients provenant d’un
centre d’oncogénétique, contre 0 % des patients provenant d’un
centre médical (9). On voit donc que les notions de sensibilité
d’un critère clinique peuvent varier de façon significative en fonc-
tion des populations étudiées.
LES CRITÈRES DE BETHESDA RÉVISÉS
Le syndrome HNPCC se caractérise par une réparation défi-
ciente de l’ADN (deficient DNA mismatch repair, ou MMR)
dans les cellules tumorales. Une instabilité des microsatellites
(MSI) dans la tumeur est toujours présente en cas de mutation
germinale des gènes MMR (MLH1, MSH2 et MSH6, notam-
ment). La stratégie diagnostique habituelle consiste donc à
analyser le matériel tumoral à la recherche d’une instabilité
des microsatellites et, si celle-ci est positive, à effectuer un
screening des gènes MMR. La fréquence de l’instabilité des
microsatellites dépend des critères cliniques utilisés pour sélec-
tionner les patients. Ainsi, on retrouve une instabilité des micro-
satellites chez 44 à 100 % des patients remplissant les critères
d’Amsterdam, chez 26 à 50 % des patients avec des cancers
colo-rectaux multiples, chez 32 % des patients présentant un
cancer colo-rectal avant l’âge de 30 ans, chez 31 à 57 % des
patients avec un cancer colo-rectal à localisation droite, et chez
5 à 20 % des patients avec un cancer colo-rectal sporadique
(10). Afin d’améliorer le rendement du test de l’instabilité des
microsatellites, des critères cliniques ont été définis afin
de sélectionner les cas les plus susceptibles de présenter une
instabilité significative (MSI-H). Il s’agit des critères dits de
Bethesda, établis par une conférence du NCI (10). Le tableauV
montre ces sept critères cliniques. Une variante de ces critères
a été définie par la Société américaine de gastro-entérologie
(AGA), où l’âge au diagnostic de cancer (colo-rectal ou de
l’endomètre) est plus élevé, soit 50 ans au lieu de 45 ans (11).
Ces critères sélectionnent environ 15 à 20 % des patients avec
un cancer colo-rectal et permettent ainsi de limiter le nombre
de cas nécessitant une recherche de MSI. Dans un travail de
Syngal, les critères de Bethesda avaient une excellente sensi-
bilité (94 %), mais une spécificité de seulement 25 %, ce qui
signifie que trois familles sur quatre ne sont pas porteuses d’une
mutation de MLH1 ou MSH2 (12).
Tableau III.
Critères d’Amsterdam II.
✓ Au moins trois apparentés avec un cancer du spectre HNPCC
(cancers du côlon, de l’endomètre, de l’uretère, des bassinets rénaux
ou de l’intestin grêle)
✓ Au moins un des cas est un apparenté du 1er degré des deux autres
✓ Au moins deux générations sont affectées
✓ Au moins un des cas a été diagnostiqué avant 50 ans
✓ Le diagnostic de polypose adénomateuse familiale (FAP) doit être
exclu
✓ Les diagnostics doivent être confirmés par un examen histologique
Adapté de Vasen et al., 1999 (6).
Tableau IV.
Sensibilité et spécificité des critères d’Amsterdam I et II.
Auteur Familles Mutations Sensibilité
Spécificité (n) (n) (%) (%)
Critères d’Amsterdam I
Nystrom-Lahti 55 36 83 73
Moslein 39 20 77 62
Wijnen 184 92 87 63
Hutter 24 14 91 69
Bapat 33 14 62 70
Wang 75 22 54 84
Liu 83 32 65 74
Scott 95 33 63 79
Bisgaard 85 31 70 79
Critères d’Amsterdam II
Syngal 70 34 78 62
Katballe 41 17 80 68
Caldes 56 30 81 58
Park 340 217 77 46
Adapté de Kievit et al. (7).
Tableau V.
Critères de Bethesda.
✓ Patient avec un cancer dans une famille remplissant les critères
d’Amsterdam I ou II
✓ Patient avec deux cancers, synchrones ou métachrones, appartenant
au spectre HNPCC*
✓ Patient avec un cancer colo-rectal et un apparenté au 1er degré avec un
cancer colo-rectal et/ou un cancer du spectre HNPCC et/ou un
adénome colo-rectal ; un de ces cancers diagnostiqué avant l’âge de
45 ans et l’adénome colique diagnostiqué avant l’âge de 40 ans
✓ Patient avec un cancer colo-rectal ou un cancer de l’endomètre
diagnostiqué avant l’âge de 45 ans
✓ Patient présentant un cancer du côlon droit avec un aspect
indifférencié (solide, cribriforme) diagnostiqué avant l’âge de 45 ans
✓ Patient présentant un cancer colo-rectal avec des cellules de type
“bague à chaton” diagnostiqué avant l’âge de 45 ans
✓ Patient avec un adénome colique diagnostiqué avant l’âge de 40 ans
* Cancer colo-rectal, de l’endomètre, de l’ovaire, de l’estomac, des voies
hépatobiliaires, de l’intestin grêle, cancer à cellules transitionnelles des bassinets
rénaux ou des uretères.
Adapté de Giardiello et al. (11).
85
La Lettre du Cancérologue - Volume XIV - n° 2 - mars-avril 2005

En 2002, une conférence du NCI a redéfini les critères de
Bethesda (tableau VI) (13). Le but de cette révision est de sim-
plifier les critères afin de les rendre plus facilement applicables
et de les élargir encore, afin de pouvoir déceler, au niveau de la
population générale, un maximum de cas. Cette nouvelle défini-
tion tient compte également de l’éventail de tous les cancers qui
ont été rapportés dans le syndrome HNPCC. Cet élargissement
des critères va forcément conduire à tester davantage de patients.
Leur sensibilité et leur spécificité n’ont pas encore été étudiées,
mais on peut s’attendre à une excellente sensibilité. Le manque
de travaux concernant ces critères explique pourquoi ils n’ont pas
été adoptés dans l’expertise récente conduite par l’INSERM (3).
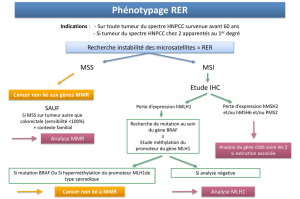
BRAF : UN COMPLÉMENT À L’ANALYSE
DES MICROSATELLITES ?
Après identification d’une instabilité des microsatellites (MSI-H)
dans la tumeur d’un patient, un screening des gènes MMR est
réalisé. Cependant, en présence d’une extinction à l’immuno-his-
tochimie du gène MLH1, dans près de 80 % des cas, l’instabilité
des microsatellites n’est pas due à une mutation germinale de
MLH1, mais à une hyperméthylation du promoteur de MLH1.
Ces cas seront donc analysés inutilement à la recherche d’une
mutation germinale de MLH1. Un nouveau marqueur a été iden-
tifié récemment et va probablement permettre de résoudre ce pro-
blème de faux positifs (tumeur MSI-H, patient MMR-).
BRAF est un gène qui code pour une protéine kinase dans la voie
de signalisation RAS/RAF/MAPK. Les cancers colo-rectaux
MSI-H d’apparence sporadique, dont l’instabilité des microsa-
tellites est probablement due à une hyperméthylation de MLH1,
présentent fréquemment une mutation V600E sur le gène BRAF.
Une équipe internationale a recherché cette mutation dans le
matériel tumoral de 206 cas de cancers colo-rectaux sporadiques
MSI-H et dans 111 cas de cancers colo-rectaux de patients por-
teurs d’une mutation de MLH1 ou MSH2 (14). Pour les cas spo-
radiques, la mutation V600E du gène BRAF a été identifiée dans
40 % des cas. En revanche, elle n’a jamais été isolée dans les
111 cas de cancers colo-rectaux de patients porteurs d’une muta-
tion de MLH1 ou MSH2. Une analyse de BRAF pourrait donc
permettre d’identifier 40 % des cas MSI-H chez qui un screening
des gènes MMR pourrait être évité.
CONCLUSION
Le syndrome HNPCC reste une pathologie largement sous-
diagnostiquée. L’adoption en pratique clinique des critères de
Bethesda révisés devrait permettre d’identifier un nombre accru
de porteurs d’une mutation d’un gène MMR. Il faudra probable-
ment attendre la publication d’études validant ces nouveaux cri-
tères avant de les voir définitivement adoptés par les différentes
sociétés d’oncogénétique. En complément de la recherche de
l’instabilité des microsatellites, l’analyse de la mutation V600E
du gène BRAF dans le matériel tumoral permettra de mieux sélec-
tionner les patients chez qui une recherche de mutation d’un gène
MMR s’avère nécessaire.
■
R
ÉFÉRENCES
B
IBLIOGRAPHIQUES
1. De la Chapelle A. Inherited human diseases: victories, challenges, disap-
pointments. Am J Hum Genet 2003;72:236-40.
2. Jarvinen HJ, Aarnio M, Mustonen H et al. Controlled 15-year trial on
screening for colorectal cancer in families with hereditary nonpolyposis colo-
rectal cancer. Gastroenterology 2000;118:829-34.
3. Olschwang S, Bonaiti C, Feingold J et al. Identification and management of
HNPCC syndrome (hereditary non polyposis colon cancer), hereditary predis-
position to colorectal and endometrial adenocarcinomas. Bull Cancer 2004;
91:303-15.
4. Vasen HF, Mecklin JP, Khan PM et al. The International Collaborative
Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis
Colon Rectum 1991;34:424-5.
5. Watson P, Lynch HT. Extracolonic cancer in hereditary nonpolyposis colo-
rectal cancer. Cancer 1993;71:677-85.
Tableau VI.
Critères de Bethesda révisés.
✓ Patient avec un cancer colo-rectal avant l’âge de 50 ans
✓ Patient avec deux cancers, synchrones ou métachrones, appartenant
au spectre HNPCC élargi*, indépendamment de l’âge
✓ Patient présentant un cancer colo-rectal avant l’âge de 60 ans avec
une histologie évoquant la présence d’une instabilité des
microsatellites**
✓ Patient présentant un cancer colo-rectal avec au moins un apparenté
du 1er degré présentant un cancer du spectre HNPCC élargi* ; un de
ces cancers diagnostiqué avant l’âge de 50 ans
✓ Patient présentant un cancer colo-rectal avec au moins deux
apparentés du 1er ou du 2edegré présentant un cancer du spectre
HNPCC élargi*, sans limitation d’âge
* Cancer colo-rectal, de l’estomac, du pancréas, de l’intestin grêle, des voies
hépatobiliaires, de l’endomètre, de l’ovaire, des bassinets rénaux ou des uretères,
du système nerveux central (glioblastome dans le syndrome de Turcot), adénome
ou carcinome des glandes sébacées (dans le syndrome de Muir-Torre).
** Tumeur avec soit : une infiltration lymphocytaire, une réaction inflammatoire
de type Crohn, une différenciation mucineuse ou en cellules de type “bague à
chaton”, un aspect de croissance médullaire.
Adapté de Umar et al. (13).
Informations disponibles sur Internet
Numéros d’accès et liens contenus dans cet article :
HNPCC France, www.hnpcc.france.free.fr/
Online Mendelian Inheritance in Man (OMIM), www.ncbi.nlm.nih.gov/
entrez/query.fcgi?db=OMIM
– BRAF : (#164757 V-RAF MURINE SARCOMA VIRAL
ONCOGENE HOMOLOG B1; BRAF)
– HNPCC : (#114500 COLORECTAL CANCER, HEREDITARY
NONPOLYPOSIS; HNPCC)
– MLH1 : (#120436 COLON CANCER, FAMILIAL
NONPOLYPOSIS, TYPE 2, MLH1)
– MSH2 : (#120435 COLON CANCER, FAMILIAL
NONPOLYPOSIS, TYPE 1, MSH2)
– MSH6 : (#600678 COLORECTAL CANCER, HEREDITARY
NONPOLYPOSIS, TYPE 5, MSH6)
LE GÈNE DU MOIS
86
La Lettre du Cancérologue - Volume XIV - n° 2 - mars-avril 2005

87
La Lettre du Cancérologue - Volume XIV - n° 2 - mars-avril 2005
6. Vasen HF, Watson P, Mecklin JP et al. New clinical criteria for hereditary
nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the
International Collaborative Group on HNPCC. Gastroenterology 1999;
116:1453-6.
7. Kievit W, de Bruin JH, Adang EM et al. Current clinical selection strategies
for identification of hereditary non-polyposis colorectal cancer families are
inadequate: a meta-analysis. Clin Genet 2004;65:308-16.
8. Wagner A, Barrows A, Wijnen JT et al. Molecular analysis of hereditary non-
polyposis colorectal cancer in the United States: high mutation detection rate
among clinically selected families and characterization of an American founder
genomic deletion of the MSH2 gene. Am J Hum Genet 2003;72:1088-100.
9. Terdiman JP, Levin TR, Allen BA et al. Hereditary nonpolyposis colorectal
cancer in young colorectal cancer patients: high-risk clinic versus population-
based registry. Gastroenterology 2002;122:940-7.
10. Rodriguez-Bigas MA, Boland CR, Hamilton SR et al. A National Cancer
Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome:
meeting highlights and Bethesda guidelines. J Natl Cancer Inst 1997;
89:1758-62.
11. Giardiello FM, Brensinger JD, Petersen GM. AGA technical review on
hereditary colorectal cancer and genetic testing. Gastroenterology 2001;
121:198-213.
12. Syngal S, Fox EA, Eng C et al. Sensitivity and specificity of clinical crite-
ria for hereditary non-polyposis colorectal cancer associated mutations in
MSH2 and MLH1. J Med Genet 2000;37:641-5.
13. Umar A, Boland CR, Terdiman JP et al. Revised Bethesda guidelines for
hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatel-
lite instability. J Natl Cancer Inst 2004;96:261-8.
14. Domingo E, Laiho P, Ollikainen M et al. BRAF screening as a low-cost
effective strategy for simplifying HNPCC genetic testing. J Med Genet 2004;
41:664-8.
15. Salovaara R, Loukola A, Kristo P et al. Population-based molecular
detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol 2000;
18:2193-200.
16. Hemminki K, Li X. Familial colorectal adenocarcinoma and hereditary
nonpolyposis colorectal cancer: a nationwide epidemiological study from Swe-
den. Br J Cancer 2001;84:969-74.
17. Katballe N, Christensen M, Wikman FP et al. Frequency of hereditary
non-polyposis colorectal cancer in Danish colorectal cancer patients. Gut
2002;50:43-51.
18. Aaltonen LA, Salovaara R, Kristo P et al. Incidence of hereditary nonpo-
lyposis colorectal cancer and the feasibility of molecular screening for the
disease. N Engl J Med 1998;338:1481-7.
1
/
4
100%