ephe_dip_sturny-leclere_2012.p

ETUDE DES SIGNAUX MACROPHAGIQUES
CELLULAIRES EN R´
EPONSE `
A L’INFECTION PAR
DIFF´
ERENTES SOUCHES CLINIQUES DE
CRYPTOCOCCUS NEOFORMANS
Aude Sturny-Lecl`ere
To cite this version:
Aude Sturny-Lecl`ere. ETUDE DES SIGNAUX MACROPHAGIQUES CELLULAIRES EN
R´
EPONSE `
A L’INFECTION PAR DIFF´
ERENTES SOUCHES CLINIQUES DE CRYPTO-
COCCUS NEOFORMANS. Biologie cellulaire. 2012. <hal-01472831>
HAL Id: hal-01472831
https://hal-ephe.archives-ouvertes.fr/hal-01472831
Submitted on 21 Feb 2017
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-
entific research documents, whether they are pub-
lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destin´ee au d´epˆot et `a la diffusion de documents
scientifiques de niveau recherche, publi´es ou non,
´emanant des ´etablissements d’enseignement et de
recherche fran¸cais ou ´etrangers, des laboratoires
publics ou priv´es.

!
"#$"!%&'()*!+*!,-'-./&012*!345!
6!
,787359:"!;"!<="83"7>8","85!3?#@:7"?:!"5!;"!<A!:"B$":B$"!
!
@BC<"!#:A57D?"!;"3!$A?5"3!@5?;"3!
!
Sciences'de'la'Vie'et'de'la'Terre'
!
MÉMOIRE'
!
0/EF*'GE!0&/!
!
!
Sturny'Leclère'Aude'
!
!
#-)/!H=-IG*'G2-'!+)!+20HJK*!+*!H=@L-H*!#/&G2()*!+*F!$&)G*F!@G)+*F!
!
!
!
ETUDE'DES'SIGNAUX'MACROPHAGIQUES'CELLULAIRES'EN'
RÉPONSE'À'L’INFECTION'PAR'DIFFÉRENTES'SOUCHES'
CLINIQUES'DE'CRYPTOCOCCUS)NEOFORMANS'
!
!
3-)G*')!H*!M6!N)2'!MO6M!
!
!
;*P&'G!H*!Q)/R!F)2P&'G!!
;/S!B&'()*!%/)'-!T!#/EF2+*'G!
;/S!"H!$&L12K2!U1&H2+!T!5)G*)/!0E+&.-.2()*!
#/S!;/-K*/!V/&'W-2F*!T!5)G*)/!FL2*'G2X2()*!
;/S!>-).*-'!,&/2*Y<2F*!T!:&00-/G*)/!
;/S!B12.'&/+!,2L1*H!T!"Z&K2'&G*)/!
4*/'*HY#&)2HH&L!V/E+E/2()*!T!"Z&K2'&G*)/!
!
!
,EK-2/*!0/E0&/E!F-)F!H&!+2/*LG2-'!+*!T!
#/S!;:C,":!V/&'W-2F*![X/&'L-2F*S+/-K*/\0&FG*)/SX/]!
?'2GE!+*!,RL-H-.2*!,-HEL)H&2/*!!B8:3!?:A!^O6M!
7'FG2G)G!#&FG*)/!Y!M_YM`!/)*!+)!;-LG*)/!:-)Z!Y!!a_aMb!#&/2F!B*+*Z!6_!
"G!+*!
;/S!"<!$AB$7,7!U1&H2+![c1&H2+S*Hd1&L12K2\)0KLSX/]!
B*'G/*!+*!/*L1*/L1*!+*!H=7'FG2G)G!+)!L*/P*&)!*G!+*!H&!K-*HH*!E02'2e/*!
B$?!#2G2E!3&H0fG/2e/*!Y!!bag!I+!+*!H=1J02G&H!Y!!a_h_6!#&/2F!B*+*Z!6^!
!

!
"#$"!%&'()*!+*!,-'-./&012*!345!
M!
@BC<"!#:A57D?"!;"3!$A?5"3!@5?;"3!
Sciences'de'la'Vie'et'de'la'Terre'
!
ETUDE'DES'SIGNAUX'MACROPHAGIQUES'CELLULAIRES'EN'RÉPONSE'À'
L’INFECTION'PAR'DIFFÉRENTES'SOUCHES'CLINIQUES'DE'
CRYPTOCOCCUS)NEOFORMANS'
!
3G)/'R!<*LHe/*!A)+*!
RÉSUMÉ'
'
Cryptococcus neoformans est une levure capsulée responsable d’infections opportunistes chez
les patients présentant un déficit immunitaire et plus particulièrement chez ceux atteints du
syndrome de l'immunodéficience acquise (SIDA). <&! X-/K*! H&! 0H)F! ./&P*! +*! H&!
L/R0G-L-LL-F*!*FG!)'*!KE'2'.-Y*'LE01&H2G*!()2!/*FG*!K-/G*HH*!+&'F!MOi!+*F!L&F!K&H./E!
)'!G/&2G*K*'G!&'G2X-'.2()*!&+&0GES!<&!L-'G&K2'&G2-'!+-''*!H2*)!j!)'*!0/2K-Y2'X*LG2-'!
0)HK-'&2/*S!<*F!K&L/-01&.*F!&HPE-H&2/*F!F-'G!H&!0/*K2e/*!L2IH*!+)! 0&G1-.e'*! *G!P-'G!
+ELH*'L1*/! H&! /E0-'F*! 2''E*S! <*! /EF*&)! LRG-c2'2()*! Q-)*! )'! /JH*! K&Q*)/! +&'F!
H=-/L1*FG/&G2-'! +=)'*! /E0-'F*! 2KK)'2G&2/*! *XX2L&L*S! ALG)*HH*K*'Gg! +*! '-KI/*)F*F!
+-''E*F!F-'G!+2F0-'2IH*F!F)/!H&!P2/)H*'L*!+*!C.#neoformansS!"'!/*P&'L1*g!H&!/E0-'F*!+*F!
K&L/-01&.*F!j!H=2'X*LG2-'!0&/!H&!H*P)/*!/*FG*!*'L-/*!j!&00/-X-'+2/S!<*F!+-''E*F!0)IH2E*F!
F)/!L*GG*!/E0-'F*!2KK)'2G&2/*!F-'G!G/eF!P&/2&IH*F!F*H-'!H*!0/-G-L-H*!*Z0E/2K*'G&HS!!
8-F!-IQ*LG2XF!EG&2*'G!+=EG)+2*/!H&!/E0-'F*!L*HH)H&2/*!+*F!K&L/-01&.*F!&0/eF!2'G*/&LG2-'!
&P*L!+*F!F-)L1*F!LH2'2()*F!2FF)*F!+*!0&G2*'GF!&GG*2'GF!+*!L/R0G-L-LL-F*S!!
A0/eF! -0G2K2F&G2-'! +=)'! 0/-G-L-H*! +=2'G*/&LG2-'! K&L/-01&.*FkH*P)/*Fg! '-)F! &P-'F!
EG)+2E! &P*L! )'! 0&'*H! +*! F-)L1*F! LH2'2()*F! 0-FFE+&'G! +*F! L&/&LGe/*F! +*! P2/)H*'L*!
+2XXE/*'GFg!H=*Z0/*FF2-'!()&'G2G&G2P*!+*F!F2.'&)Z!K&L/-01&.2()*F![58VYαg!7<Yhg!7<Y6Og!7<Y
6βg!,&''-F*!/EL*0G*)/g!5<:bg!28C3]!0&/!H&!G*L1'2()*!+*!#B:!*'!G*K0F!/E*HS!!
A)!G/&P*/F!+*!H&!+2P*/F2GE!+*!L*F!F-)L1*F!LH2'2()*Fg!'-)F!&P-'F!-IF*/PE!H&!+2P*/F2GE!+*!H&!
/E0-'F*!+*!H=1JG*g!+*F!K*FF&.*/F!2KK)'2G&2/*F!EG&2*'G!F0EL2X2()*K*'G!+E/E.)HEF!F*H-'!
H*F!F-)L1*FS!!
#H)F2*)/F! K*FF&.*/F! LHEF! +*! H&! /E0-'F*! 2'XH&KK&G-2/*! F-'G! *'L-/*! *'! L-)/F! +=EG)+*S!
A0/eF!&'&HRF*!()&'G2G&G2P*!+*F!/EF)HG&GF!0/EH2K2'&2/*Fg!'-)F!*FF&R*/-'F!+*!+EG*/K2'*/!F2!
H=)'! +*! L*F! K*FF&.*/F! 0-FFe+*! H*! 0-G*'G2*H! +*! l!K&/()*)/! +*! 0/-'-FG2L!m! '-)F!
0*/K*GG&'G! +*! F)2P/*! H=EP-H)G2-'! +*! H=2'X*LG2-'! *G! &2'F2! +=&00/E1*'+*/! H*F! L&F! H*F! 0H)F!
FEPe/*F!+*!L*GG*!K&H&+2*S!
!
,C53YB<@3!T!Cryptococcus#neoformansg!K&L/-01&.*g!LRG-c2'*Fg!#B:!G*K0F!/E*Hg!
*Z0/*FF2-'!()&'G2G&G2P*!
!
!
!

!
"#$"!%&'()*!+*!,-'-./&012*!345!
^!
RESUME'.............................................................................................................................................................'2!
INTRODUCTION'..............................................................................................................................................'4!
7S!B:n#5CBCBB?3!8"CVC:,A83!T!<"!B$A,#7>8C8!SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS!_!
I.1.#Généralités#.................................................................................................................................................................#5!
I.2.#Répartition#géographique#et#habitats#...........................................................................................................#5!
I.3.#Facteurs#de#virulence#............................................................................................................................................#6!
77S!!<A!B:n#5CBCBBC3"!SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS!a!
II.1.#Epidémiologie#.........................................................................................................................................................#7!
II.2.#Clinique/Pronostic#................................................................................................................................................#8!
II.3.#Physiopathologie#...................................................................................................................................................#9!
777S!<A!:"#C83"!7,,?875A7:"!SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS!6O!
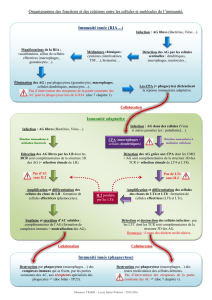
III.1.#Immunité#innée#..................................................................................................................................................#10!
III.2.#Immunité#adaptative#.......................................................................................................................................#14!
III.2.A.#Réponse#immunitaire#à#médiation#cellulaire#....................................................................................#14!
III.2.B.#Réponse#immunitaire#à#médiation#humorale#...................................................................................#15!
III.2.C.#Les#cytokiniques#et#les#chimiokines#........................................................................................................#16!
74S!:"#C83"!7,,?875A7:"!A!<=78V"B57C8!#A:!C.#NEOFORMANS!SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS!6a!
IV.1.#La#réponse#immunitaire#innée#.....................................................................................................................#18!
IV.2.#La#réponse#immunitaire#adaptatrice#........................................................................................................#20!
4S!C?57<3!;="5?;"3!SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS!M6!
V.1.#Les#modèles#d’études#de#la#réponse#immunitaire#..................................................................................#21!
V.2.#Etude#de#l’expression#par#PCR#en#temps#réel#...........................................................................................#25!
47S!#:C%<",A57D?"!"5?;7""!"5!C%N"B57V3!SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS!!
MATERIEL'............................................................................................................................................................''
&'METHODES'......................................................................................................................................................'!
7S!3C?B$"3!;"!C.#NEOFORMANS!SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS!!
I.1.#Origine#............................................................................................................................................................................##
I.2.#Repiquage,#stockage#.................................................................................................................................................##
I.3.#Préculture#et#préparation#des#levures#avant#contact#avec#les#macrophages#.................................##
II.#Matériel#et#culture#cellulaire#..................................................................................................................................##
II.1.#Source#............................................................................................................................................................................#!
II.2.#Entretien#......................................................................................................................................................................#!
II.3.#Préparation#avant#contact#avec#les#levures#.................................................................................................#!
777S!,C;"<"!;=785":AB57C8!,AB:C#$A>"3kC.#NEOFORMANS!SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS!!
III.1.#Anticorps#monoclonal#IgG1#E1#.........................................................................................................................##
III.2.#Interaction#macrophages/C.#neoformans#...................................................................................................##
III.3.#Optimisation#du#modèle#......................................................................................................................................#!
74S!#:"#A:A57C8!;"3!A:8!SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS!!
IV.1.#Protocole#d’extraction#d’ARN#............................................................................................................................##
IV.2.#Traitement#DNase#..................................................................................................................................................#!
IV.3.#Synthèse#de#cDNA#...................................................................................................................................................#!
4S!#B:!D?A8575A574"!SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS!!
V.1.#Light#Cycler#480#II#Roche#......................................................................................................................................#!
V.2.#Dessin#des#amorces#..................................................................................................................................................#!
V.3.#Protocole#d’amplification#par#PCR#...................................................................................................................#!
V.4.#Mise#au#point#du#protocole#de#qPCR#................................................................................................................#!
V.5.#Critères#d’analyse#des#données#générées#par#qPCR#...................................................................................##
V.6.#Électrophorèse#...........................................................................................................................................................#!
V.7.#Mise#au#point#des#courbes#standards#de#référence#....................................................................................#!
V.8.#Détails#de#la#méthode#de#calcul#de#quantification#relative#par#le#
ΔΔ
dt#...........................................#!
RESULTATS'.........................................................................................................................................................'!
7S!C#57,73A57C8!;?!,C;"<"!B"<<?<A7:"!"o#":7,"85A<!SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS!!

!
"#$"!%&'()*!+*!,-'-./&012*!345!
b!
77S!C#57,73A57C8!;"3!#:C5CBC<"3!;"!D?A857V7BA57C8!;"!<="o#:"337C8!SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS!!
II.1#Mise#au#point#et#validation#du#protocole#d’extraction#d’ARN#...............................................................##
II.2.#Intérêt#du#traitement#à#la#DNase#..#II.3#Mise#au#point#et#validation#du#protocole#de#synthèse#
de#ADNcII.4.#Mise#au#point#du#protocole#de#PCR,#validation#des#couples#d’amorces#et#validation#
des#critères#d’analyse#:#Exemple#pour#le#gène#de#la#
β
^Actine#.........................................................................#!
II.5.#Validation#des#courbes#standard#de#référence#pour#tous#les#gènes#cibles#:#Exemple#pour#le#
gène#de#l’IL^6#.......................................................................................................................................................................##
II.6.#Démarche#de#reproductibilité#des#courbes#standard#...............................................................................#!
777S!A8A<n3"!;="o#:"337C8!;"3!>"8"3!SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS!!
III.1.#Validation#de#la#méthode#de#quantification#et#du#système#..................................................................#!
III.2.#Résultats#du#niveau#d’expression#des#gènes#sur#une#cinétique#précoce#..........................................#!
III.3.#Résultats#d’expression#des#gènes#liées#aux#conditions#d’opsonisation#............................................##
par#E1#.....................................................................................................................................................................................#!
DISCUSSION'........................................................................................................................................................'!
&'PERSPECTIVES'...............................................................................................................................................'!
BIBLIOGRAPHIE'................................................................................................................................................'!
ANNEXES'..............................................................................................................................................................'!
'
BIBLIOGRAPHIE'...........................................................................................................................................'32!
!
'
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
1
/
42
100%