A RANDOMIZED DOUBLE-BLIND, PLACEBO CONTROLLED STUDY OF IN

Etude MONALEESA-3, v 0.0 du 09.03.2015 dngn 24.02.2016
A RANDOMIZED DOUBLE-BLIND, PLACEBO CONTROLLED STUDY OF RIBOCICLIB IN
COMBINATION WITH FULVESTRANT FOR THE TREATMENT OF POSTMENOPAUSAL
WOMEN WITH HORMONE RECEPTOR POSITIVE, HER2-NEGATIVE, ADVANCED
BREAST CANCER WHO HAVE RECEIVED NO OR ONLY ONE LINE OF PRIOR
ENDOCRINE TREATMENT
“Etude MONALEESA-3”
Sponsor: Novartis
CONTACTS AUX HUG :
Médecin chef de clinique: Dr Noémie Lang, 079 553 23 98
Investigateur responsable: Dr Alexandre Bodmer, 079 55 32 405
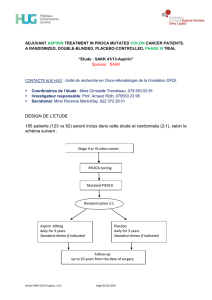
DESIGN DE L’ETUDE
Environ 660 femmes, dans 300 centres d’environ 30 pays différents seront randomisées
selon un ratio 2:1 dans l’un des deux groupes de traitement, selon le schéma suivant :

Etude MONALEESA-3, v 0.0 du 09.03.2015 dngn 24.02.2016
OBJECTIF DE L’ETUDE
L’objet de cette étude est de découvrir si le ribociclib (LEE011), lorsqu’on l’ajoute au
traitement disponible dans le commerce, le fulvestrant (Faslodex®), est sûr et s’il contribue à
réduire la croissance des cellules cancéreuses chez les femmes en post-ménopause ayant
un cancer du sein à récepteurs hormonaux positifs, HER-2 négatif, au stade avancé, qui
n’ont reçu aucun ou une seule ligne de traitement endocrinien auparavant.
CRITERES D’ELIGIBILITE
#
INCLUSION CRITERIA
1
Patient is an adult ≥ 18 years old at the time of informed consent and has signed informed consent before any trial
related activities and according to local guidelines.
2
Patient is postmenopausal. Postmenopausal status is defined either by:
Prior bilateral oophorectomy
Age ≥60
Age <60 and amenorrheic for 12 or more months in the absence of chemotherapy, tamoxifen, toremifene,
or ovarian suppression and FSH and estradiol in the postmenopausal range per local normal range
Note: For women with therapy-induced amenorrhea, , serial measurements of FSH and/or estradiol are needed to ensure
postmenopausal status (NCCN 2.2015). Ovarian radiation or treatment with a luteinizing hormone-releasing hormone agonist
(LH-RHa) (goserelin acetate or leuprolide acetate) is not permitted for induction of ovarian suppression in this trial.
3
Patient has a histologically and/or cytologically confirmed diagnosis of estrogen-receptor positive and/or
progesterone receptor positive breast cancer by local laboratory.
4
Patient has HER2-negative breast cancer defined as a negative in situ hybridization test or an IHC status of 0, 1+
or 2+. If IHC is 2+, a negative in situ hybridization (FISH, CISH, or SISH) test is required by local laboratory
testing
5
Patient must have either:
Measurable disease, i.e., at least one measurable lesion as per RECIST 1.1. (a lesion at a previously
irradiated site may only be counted as a target lesion if there is a clear sign of progression since the
irradiation).
OR
If no measurable disease is present, then at least one predominantly lytic bone lesion must be present
(patients with no measurable disease and only one predominantly lytic bone lesion that has been
previously irradiated are eligible if there is documented evidence of disease progression of the bone
lesion after irradiation).
6
Patient has advanced (loco regionally recurrent not amenable to curative therapy or metastatic) breast cancer.
Patients may be :
newly diagnosed advanced breast cancer, treatment naïve
relapsed with documented evidence of progression more than 12 months from completion of
(neo)adjuvant endocrine therapy with no treatment for metastatic disease
relapsed with documented evidence of progression on or within 12 months from completion of
(neo)adjuvant endocrine therapy with no treatment for metastatic disease
relapsed with documented evidence of progression more than 12 months from completion of adjuvant
endocrine therapy and then subsequently progressed with documented evidence of progression after one
line of endocrine therapy (with either an antiestrogen or an aromatase inhibitor) for metastatic disease
newly diagnosed advanced breast cancer, then progressed with documented evidence of progression
after one line of endocrine therapy (with either an antiestrogen or an aromatase inhibitor)
Note: Patient who relapsed with documented evidence of relapse on/or within 12 months from completion of adjuvant
endocrine therapy and then subsequently progressed with documented evidence of progression after one line of endocrine
therapy (with either an antiestrogen or an aromatase inhibitor) for metastatic disease will NOT be included in the study.
7
Patient has an Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1
8
Patient has adequate bone marrow and organ
function as defined by the following laboratory
values (as assessed by central laboratory for
eligibility):
Absolute neutrophil count ≥ 1.5 × 109/L
Platelets ≥ 100 × 109/L
Hemoglobin ≥ 9.0 g/dL
INR ≤1.5
Serum creatinine < 1.5 mg/dL

Etude MONALEESA-3, v 0.0 du 09.03.2015 dngn 24.02.2016
Total bilirubin < ULN except for patients with Gilbert’s
syndrome who may only be included if the total bilirubin is ≤
3.0 × ULN or direct bilirubin ≤ 1.5 × ULN
AST < 2.5 × ULN, except for patients with liver metastasis,
who are only included if the AST is < 5 × ULN
ALT< 2.5 × ULN, except for patients with liver metastasis,
who are only included if the AST is < 5 × ULN
Patient must have the following laboratory values within
normal limits or corrected to
within normal limits with supplements before the first dose
of study medication:
Sodium
Potassium
Magnesium
Phosphorus
Total Calcium (corrected for serum albumin)
#
EXCLUSION CRITERIA
1
Patient with symptomatic visceral disease or any disease burden that makes the patient ineligible for endocrine
therapy per the investigator’s best judgment
2
Patient has received prior treatment with chemotherapy (except for neoadjuvant/ adjuvant chemotherapy),
fulvestrant or any CDK4/6 inhibitor
3
Patients has received prior neoadjuvant/adjuvant treatment with antracyclines at cumulative doses of 450mg/m²
or more for doxorubicin or 900 mg/m² or more for epirubicin.
4
Patient with a known hypersensitivity to any of the excipients of ribociclib or fulvestrant
5
Patient with inflammatory breast cancer at screening
6
Patient is concurrently using other anti-cancer therapy
7
Patient has had major surgery within 14 days prior to starting study drug or has not recovered from major side
effects
8
Patients with Child pugh score B or C
9
Patient is currently receiving warfarin or other coumarin derived anti-coagulant, for treatment, prophylaxis or
otherwise. Therapy with heparin, low molecular weight heparin (LMWH), or fondaparinux is allowed
10
Patient has not recovered from all toxicities related to prior anticancer therapies to NCI CTCAE version 4.03
Grade ≤1. Exception to this criterion: patients with any grade of alopecia are allowed to enter the study.
11
Patient has received radiotherapy ≤ 4 weeks or limited field radiation for palliation ≤ 2 weeks prior to
randomization, and who has not recovered to grade 1 or better from related side effects of such therapy (with
the exception of alopecia) and/or from whom ≥ 25% (Ellis 1961) of the bone marrow was irradiated
12
Patient has a concurrent malignancy or malignancy within 3 years of randomization, with the exception of
adequately treated, basal or squamous cell carcinoma, non-melanomatous skin cancer or curatively resected
cervical cancer
13
Patients with central nervous system (CNS) involvement unless they meet ALL of the following criteria:
At least 4 weeks from prior therapy completion (including radiation and/or surgery) to starting the study
treatment.
Clinically stable CNS tumor at the time of screening and not receiving steroids and/or enzyme inducing
anti-epileptic medications for brain metastases.
14
Patient has impairment of gastrointestinal (GI) function or GI disease that may significantly alter the absorption
of the study drugs (e.g., ulcerative diseases, uncontrolled nausea, vomiting, diarrhea, malabsorption syndrome,
or small bowel resection).
15
Patient has a known history of HIV infection (testing not mandatory)
16
Patient has any other concurrent severe and/or uncontrolled medical condition that would, in the investigator’s
judgment, cause unacceptable safety risks, contraindicate patient participation in the clinical study or
compromise compliance with the protocol: (e.g. chronic pancreatitis, chronic active hepatitis, active untreated or
uncontrolled fungal, bacterial or viral infections, etc.)
17
Clinically significant, uncontrolled heart disease and/or recent cardiac events including any of the following:
History of angina pectoris, symptomatic pericarditis, or myocardial infarction within 12 months prior to
study entry

Etude MONALEESA-3, v 0.0 du 09.03.2015 dngn 24.02.2016
History of documented congestive heart failure (New York Heart Association functional classification III-
IV)
Documented cardiomyopathy
Left Ventricular Ejection Fraction (LVEF) < 50% as determined by Multiple Gated acquisition (MUGA)
scan or echocardiogram (ECHO)
History of any cardiac arrhythmias, e.g., ventricular, supraventricular, nodal arrhythmias, or conduction
abnormality in the previous 12 months.
Congenital long QT syndrome or family history of long QT syndrome
Systolic Blood Pressure (SBP) >160 or <90 mmHg
Bradycardia (heart rate < 50 at rest), by ECG or pulse.
On screening, inability to determine the QTcF interval on the ECG (ie: unreadable or not interpretable)
or QTcF >450 msec (using Fridericia’s correction). All as determined by screening ECG (mean of triplicate
ECGs).
18
Patient is currently receiving any of the following substances and cannot be discontinued 7 days prior to Cycle 1
Day 1:
Known strong inducers or inhibitors of CYP3A4/5, including grapefruit, grapefruit hybrids, pummelos,
star-fruit, and Seville oranges.
That have a known risk to prolong the QT interval or induce Torsades de Pointes.
That have a narrow therapeutic window and are predominantly metabolized through CYP3A4/5.
Herbal preparations/medications, dietary supplements.
19
Patient is currently receiving or has received systemic corticosteroids ≤ 2 weeks prior to starting study drug, or
who have not fully recovered from side effects of such treatment.
Note: The following uses of corticosteroids are permitted: single doses, topical applications (e.g., for rash), inhaled sprays
(e.g., for obstructive airways diseases), eye drops or local injections (e.g., intra-articular)
20
Participation in a prior investigational study within 30 days prior to enrollment or within 5-half lives of the
investigational product, whichever is longer
21
Not able to understand and to comply with study instructions and requirements.
1
/
4
100%