PREVENTION OF SYMPTOMATIC WITH – A NONINFERIORITY

Etude SAKK 96/12 dngn 02.06.2015
PREVENTION OF SYMPTOMATIC SKELETAL EVENTS WITH DENOSUMAB
ADMINISTERED EVERY 4 WEEKS VERSUS EVERY 12 WEEKS – A NONINFERIORITY
PHASE III TRIAL
“Etude SAKK 96/12”
Sponsor : SAKK
CONTACTS AUX HUG : Centre du sein
Coordinatrice de l’étude : Mme Virginie Nepote, 079 55 34 031 (sein) / Mme Delphine
Gani, 079 55 32 426 (prostate)
Investigateur responsable: Dr Alexandre Bodmer, 079 55 32 405 (sein) / Prof. Arnaud
Roth, 079 55 32 395 (prostate)
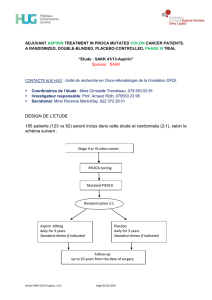
DESIGN DE L’ETUDE
1'380 patients atteints d’un cancer de la prostate au du sein avec des métastases
osseuses seront inclus dans un des deux bras suivants :
OBJECTIF DE L’ETUDE
L’objectif principal est d’établir que le traitement par dénosumab 120mg donné toutes
les 12 semaines est non-inférieur au traitement par dénosumab 120mg donné toutes
les 4 semaines.
Métastases
osseuses d'un
cancer de la
prostate résistant
à la castration ou
cancer du sein
Stratifié selon:
type de cancer
fonction rénale
état de santé selon
classification OMS
pays
Bras A (bras standard) :
denosumab 120 mg (Xgeva®) sc. q4s
Bras B (bras réduit):
3x denosumab 120 mg (Xgeva®) sc.
q4s, puis denosumab 120 mg
(Xgeva®) sc. q12s
Complémentation avec 500 mg
calcium et 400 U vitamine D par jour

Etude SAKK 96/12 dngn 02.06.2015
CRITERES D’INCLUSION / EXCLUSION
#
INCLUSION CRITERIA
1
Patient has given written informed consent
2
For breast cancer patients: Histologically confirmed diagnosis before randomization
3
For prostate cancer patients: Histologically or cytologically confirmed diagnosis before randomization
4
Patient has metastatic breast cancer (stage IV, all subtypes allowed, except small cells) or prostate cancer
(stage IV, exclude small cells) and bone metastases and is planned to receive or is receiving antineoplastic
treatment
5
Patients with prostate cancer must have evidence of disease progression on continuous androgen deprivation
therapy (CRPC)
6
Patients must have ≥ 3 bone metastases (lytic or blastic or mixed). The lesions must be documented by
radiological evaluation within 12 weeks before randomization (by X-Ray, CT scan, PET-CT, MRI scan or bone
scintigraphy)
7
WHO performance status 0-2 (see Appendix 3)
8
Age ≥ 18 years
9
Corrected serum calcium ≥ 2 mmol/l and ≤ 3 mmol/l (medical treatments to obtain serum calcium levels in the
normal range are allowed, as far as no bisphosphonates or denosumab are used)
10
Liver transaminases within normal range or not more than 3 x ULN with liver metastases. Serum total bilirubin ≤
1.5 x ULN (≤ 2.0 x ULN in case of known Gilbert’s disease)
11
Women are not breastfeeding. Women with child-bearing potential are using effective contraception (see 9.4),
are not pregnant and agree not to become pregnant during participation in the trial and during the 12 months
thereafter. A negative pregnancy test before inclusion (within 7 days) into the trial is required for all women with
child-bearing potential
12
Men agree not to father a child during participation in the trial and during 12 months thereafter
#
EXCLUSION CRITERIA
1
Definite contraindication for denosumab (e.g. hypocalcemia [Albumin-corrected serum calcium < 2.0 mmol/l])
2
History or current evidence of osteonecrosis of the jaw
3
Non-healed mucosa in oral cavity (by surgery or as a side effect of any other treatment)
4
Jaw or dental conditions that require oral surgery or if surgery or invasive dental procedures are planned
5
Prior use of denosumab for bone metastases (dose 120 mg every 4 weeks) or bisphosphonates to treat bone
metastases. Patients treated with denosumab or bisphosphonates against osteopenia or osteoporosis are
allowed to enter the trial if the last dose was more than 28 days before randomization
6
Patients with known osteoporosis (T-score ≤ -2.5) at study entry (since fractures from osteoporosis are difficult
to be discriminated from fractures through bone metastases)
7
Radiotherapy or surgery to the bone within the last two weeks before randomization or planned within 6 weeks
after randomization
8
Presence or history of CNS metastases or leptomeningeal disease. A MRI evaluation within 12 weeks before
randomization must be performed in case of suspicious symptoms
9
Psychiatric disorder precluding understanding of information on trial related topics, giving informed consent,
filling out QoL forms
10
Concurrent treatment in a clinical trial with SSE or SRE as primary endpoint
11
Known hypersensitivity to trial drug or hypersensitivity to any other component of the trial
drug (e.g. fructose)
12
Any concomitant drugs contraindicated for use with the trial drugs according to the approved product
information
13
Any psychological, familial, sociological or geographical condition potentially hampering compliance with the trial

Etude SAKK 96/12 dngn 02.06.2015
protocol
1
/
3
100%