Télécharger cette fiche au format PDF

1021
CHAPITRE G
THERAPIE GENIQUE
PRINCIPES et APPLICATIONS
EN UROLOGIE
" Si toute invention physique ou chimique est un blasphème, toute invention biolo-
gique est une perversion…
`il n’en est guère qui, à l’origine, n’ait semblé indécente ou hors nature "
J.B.S Haldane

1022
1. THÉRAPIES GÉNIQUES SOMATIQUE ET GERMI-
NALE
2. STRATÉGIES DE THÉRAPIE GÉNIQUE
3. THÉRAPIES GÉNIQUES IN VIVO ET EX VIVO
4. VECTEURS DE THÉRAPIE GÉNIQUE
5. MÉTHODES DE DÉLIVRANCE
1. STRATÉGIE CORRECTIVE ET PROTECTRICE
2. STRATÉGIE CYTOTOXIQUE ET SUICIDE
3. STRATÉGIE ANTI-SENS
4. STRATÉGIE D’IMMUNOMODULATRICE
1. STRATÉGIE CORRECTIVE
2. STRATÉGIE CYTOTOXIQUE
3. STRATÉGIE D’IMMUNOMODULATRICE
1. STRATÉGIE CORRECTIVE
2. STRATÉGIE CYTOTOXIQUE
3. STRATÉGIE ANTI-SENS
4. STRATÉGIE D’IMMUNOMODULATRICE
1. STRATÉGIE CORRECTIVE
2. STRATÉGIE CYTOTOXIQUE
3. STRATÉGIE ANTI-SENS
4. STRATÉGIE IMMUNOMODULATRICE
1. POLYKYSTOSE RÉNALE
2. GLOMÉRULONÉPHRITES
3. INSUFFISANCE RÉNALE CHRONIQUE
E. CONCLUSION
D. TRANSPLANTATION
III. STENOSES URETERALE ET
URETHRALE
II. IMPUISSANCE
I. PATHOLOGIE RENALE
C.UTILISATION EN
PATHOLOGIE BENIGNE
V. UTILISATION EN PATHOLOGIE
RENALE
IV. UTILISATION EN PATHOLOGIE
TESTICULAIRE
III. UTILISATION EN PATHOLOGIE
VESICALE
II. UTILISATION EN PATHOLOGIE
PROSTATIQUE
I. STRATEGIES THERAPEUTIQUES
B. APPLICATIONS DE LA
THERAPIE GENIQUE A
LA CANCEROLOGIE
UROLOGIQUE
II. MODALITES DE LA THERAPIE
GENIQUE
I. DEFINITION
A. PRINCIPES DE THERAPIE
GENIQUE
INTRODUCTION
PLAN

1023
THERAPIE GENIQUE
PRINCIPES et APPLICATIONS
en UROLOGIE
¨Si l’on considère le but d’un médicament comme
la restitution d’une fonction part i c u l i è re du
corps, alors l’ADN doit être tenu comme le médi-
cament absolu.¨ [1]. Cet aphorisme a guidé le for-
midable développement de la thérapie génique, dont
les champs d’application ne cessent de s’étendre et
les protocoles cliniques de se multiplier. La thérapie
génique est en passe actuellement de se constituer en
spécialité à part entière. C’est en 1944 qu’eurent lieu
les premières expériences de transfert de gènes
(transduction), avec la transformation de bactéries
par introduction d’un gène hétérologue. En 1968, les
premières cellules de mammifères furent transfor-
mées par des gènes viraux alors qu’un congrès de
thérapie génique consacré aux maladies génétiques
(hémoglobinopathies et maladie de LESCH-
NYHAN) se tenait dès 1971. En 1990, la thérapie
génique acquit sa popularité auprès des médias géné-
ralistes avec le premier essai clinique pratiqué chez
l’Homme, sous la coordination de W.F. ANDER-
SON [2]. A ce jour, près de 400 protocoles cliniques
sont en cours, incluant plus de 3500 patients à tra-
vers le monde. Ces travaux ont mobilisé, unique-
ment aux Etats-Unis, 200 millions de dollars du sec-
teur public et une somme équivalente du secteur
privé [3].
Après en avoir rappelé les principes, nous passerons
en revue les principales applications de la thérapie
génique aux domaines de l’urologie.
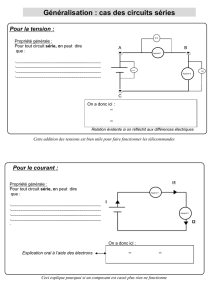
La thérapie génique est basée sur le transfert de
gène.Cette technique consiste en l’introduction dans
une cellule, dite cible, d’un matériel génétique
constitué d’un gène sous la dépendance d’une
séquence de régulation. Cette séquence de régulation
est habituellement un promoteur. Le gène est intro-
duit soit directement sous forme d’ADN nu, soit
grâce à un vecteur, dans le génome duquel il est
généralement inséré. L’expression de ce gène modi-
fiera les propriétés fonctionnelles de la cellule, appe-
lée alors cellule transduite. Cette expression, soit
restaurera une fonction perdue ou altérée, soit lui
fera acquérir une fonction nouvelle.
1. THÉRAPIES GÉNIQUES SOMATIQUE ET GERMI-
NALE
L’attitude encore prévalente considère la thérapie
génique somatique (le transfert de gène au seul
patient et non à sa descendance) comme un mode
II. MODALITES DE LA THERAPIE
GENIQUE
I. DEFINITION (FIG. 1)
A. PRINCIPES DE THERAPIE
GENIQUE
INTRODUCTION

thérapeutique classique, dont il convient de détermi-
ner le rapport entre ses risques et ses bénéfices. La
thérapie génique germinale ou embryonnaire n’est
pas encore validée, pour des considérations essen-
tiellement éthiques. Toutefois, une demande récente
d’approbation d’un protocole de thérapie génique
fœtale humaine, aux U.S.A., risque de remettre en
cause ce principe de précaution [4]. Ce protocole,
dont le promoteur est cette fois encore W. F.
ANDERSON, a pour but de soigner l’alpha-thalas-
sémie et surtout le SCID (déficit immunitaire com-
biné par déficit en enzyme ADA), lequel a fait l’ob-
jet du premier protocole d’essai clinique humain.
2. STRATÉGIES DE THÉRAPIE GÉNIQUE
La thérapie génique consiste en la modification
fonctionnelle d’une cellule, et certaines de ses
applications sont d’une mise au point particulière-
ment délicate. Lorsque l’anomalie à corriger touche
des cellules hautement différenciées, elles sont les
seules cibles possibles. Par exemple, la correction de
la ß-thalassémie ne s’entend que par l’implantation
du gène de la ß-globine dans les pro-géniteurs
hématopoïétiques. Il est des cas plus simples, où la
fonction cellulaire est moins spécialisée. Si la protéi-
ne anormale est, par exemple, sécrétée, son anoma-
lie peut alors être corrigée à distance (c’est le cas des
maladies lysosomales [5]).
Ainsi deux types de protocoles peuvent-ils être gros-
sièrement individualisés.
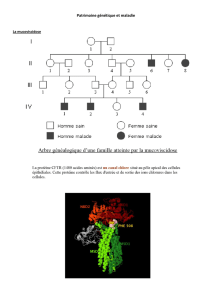
a) Protocoles de marquage cellulaire (Fig.2)
De tels protocoles permettent, soit de suivre le deve-
nir d’une cellule et de sa descendance, soit d’évaluer
une voie d’administration, un vecteur ou la durée
d’expression du transgène. Il s’agit donc de proto-
coles de compréhension, qui, in vivo, s’inscrivent uni-
quement dans le cadre d’essais de phase I. Il ne s’agit
aucunement de protocoles à visée thérapeutique [6].
Ils consistent à insérer un gène hétérologue, dit gène
rapporteur, dans le génome de la cellule cible. La
traduction de ce gène, en une protéine facilement
repérable, marque la cellule transduite, “l’estam-
pille” en quelque sorte, et permet de la détecter spé-
cifiquement dans le temps ou l’espace. Outre les
gènes de la luciférase et de la «green fluorescent pro-
tein» (GFP), le gène le plus couramment utilisé est
lac-Z, le gène de la ß-galactosidase d’escherichia
1024
Figure 1: Les acteurs de la Thérapie Génique: la thérapie génique suppose le transfert d’un gène dans une cellule.
Ce gène sera alors exprimé dans la cellule transduite

1025
Figure 2 : Essai de thérapie génique en utilisant un gène rapporteur, la ß-galactosidase. Le marquage peut être spécifique des
cellules épithéliales prostatiques (1 à 4) ou des fibroblastes. (5).
12
3 4
5
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
1
/
32
100%