Syndrome de Cogan

CAS CLINIQUE
22 | La Lettre d’ORL et de chirurgie cervico-faciale • n° 330 - juillet-août-septembre 2012
Mots-clés :
Surdité brusque– Auto-immunité– Corticothérapie.
Keywords:
Sudden hearing loss– Autoimmunity– Steroids.
Syndrome de Cogan
Cogan’s syndrome: case report
D. Bouccara*
* Service d’ORL, hôpital Beaujon, Clichy.
Q
uand suspecter une maladie de système devant une
surdité de perception ? Quels sont les arguments qui
vont faire suspecter une atteinte auto-immune de
l’oreille interne ? Ces situations sont relativement rares mais
méritent un diagnostic précoce. En effet, les signes auditifs
sont parfois les premiers, et l’évolution peut se compliquer
de l’atteinte d’autres organes : rein, cœur... Le rôle du médecin
ORL est donc d’évoquer le diagnostic et de le confronter à
l’avis du médecin interniste, qui décidera du bilan adapté et
des modalités thérapeutiques. Le syndrome de Cogan illustre
cette approche pluridisciplinaire.
Observation
Un patient âgé de 23 ans consulte pour une surdité brusque
gauche. Il signale comme seul antécédent des bronchites non
compliquées durant l’enfance. Il n’y a pas de notion d’allergie,
pas d’atteinte auditive connue, pas de traumatisme ou d’expo-
sition au bruit. Quelques semaines auparavant, il a été traité
par un collyre pour une atteinte oculaire bilatérale – selon
lui, une conjonctivite. Il signale qu’il a persisté une rougeur
oculaire durant plusieurs semaines malgré le traitement. Très
récemment, il a présenté une baisse brutale de l’audition à
gauche, sans facteur déclenchant, associée à des acouphènes
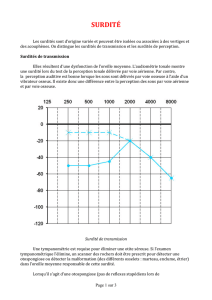
droits. L’examen ORL est normal. L’audiogramme retrouve
une surdité de perception gauche (figure 1). Le diagnostic de
surdité brusque “idiopathique” est retenu. Le patient reçoit un
traitement associant des corticoïdes et des vasodilatateurs.
L’évolution est marquée par une récupération quasi complète
de l’audition. L’IRM qui est réalisée montre l’absence de lésion
évolutive.
Quelques jours après l’arrêt du traitement, le patient présente
d’une part une récidive de l’atteinte auditive gauche, d’autre
part, une rougeur oculaire. L’examen ophtalmologique retrouve
une kératite interstitielle, et le diagnostic de syndrome de Cogan
est établi. Un complément de bilan est effectué en médecine
interne avant instauration d’une corticothérapie au long cours. Malgré
ce traitement, l’évolution est marquée par une dégradation auditive
motivant le recours aux traitements immunosuppresseurs (métho-
trexate et azathioprine), qui permettront de stabiliser l’audition.
Discussion
Le syndrome de Cogan est une vascularite rare caractérisée par l’asso-
ciation d’une atteinte ophtalmologique et d’une atteinte cochléo-
vestibulaire (1, 2). Cette affection débute généralement entre 20 et
30 ans. Différentes hypothèses étiopathogéniques ont été avancées,
en particulier infectieuse.
L’atteinte oculaire est habituellement inaugurale, mais elle peut être
méconnue si elle est transitoire. Il s’agit d’une kératite interstitielle
bilatérale entraînant des douleurs oculaires, un larmoiement et une
photophobie. Lors de l’examen ophtalmologique, l’aspect à la lampe
à fente est voisin de celui des kératites infectieuses virales et chlamy-
diennes, ce qui peut retarder le diagnostic. D’autres atteintes oculaires
sont possibles au cours du syndrome de Cogan : kératite superficielle,
conjonctivite, choroïdite, uvéite antérieure, sclérite et épisclérite. Le
traitement de ces atteintes oculaires est fondé sur l’administration
de corticoïdes locaux.
Les signes cochléo-vestibulaires apparaissent le plus souvent après
les signes ophtalmologiques, avec un intervalle atteignant parfois
plusieurs mois (entre 3 et 6 mois habituellement). L’atteinte débute
brusquement et associe une hypoacousie avec acouphènes à des signes
vestibulaires proches de ceux rencontrés au cours de la maladie de
Ménière. La surdité étant souvent brutale, elle motive un traitement
corticoïde, tel que proposé lors des surdités brusques. En général, il
existe une réponse franche à ce traitement, avec récupération de
l’audition au terme d’un traitement corticoïde de l’ordre d’une semaine.
À l’arrêt de la corticothérapie, la surdité récidive habituellement en
quelques jours, ce qui fait reprendre ce traitement.
Plusieurs diagnostics sont alors évoqués :
➤
surdité fluctuante telle que rencontrée lors des pathologies
pressionnelles (maladie de Ménière débutante), mais l’atteinte auditive
touche habituellement prioritairement les fréquences graves ;

CAS CLINIQUE
Figure 1. Examen audiométrique tonal et vocal retrouvant une surdité de perception unilatérale gauche prédominant sur les fréquences aiguës.
Examen audiométrique tonal
Gauche (kHz)
dB dB
75 75 75 75 75 75 75 75
0,125 0,250 0,5 0,75 1 1,5 2 3 4 6 8 12 16
– 20
– 10
0
10
20
30
40
50
60
70
80
90
100
110
120
AC
BC
FF
Droite (kHz)
0 0 0 0 0 0 0 0
0,125 0,250 0,5 0,75 1 1,5 2 3 4 6 8 12 16
– 20
– 10
0
10
20
30
40
50
60
70
80
90
100
110
120
AC
BC
FF
Examen audiométrique vocal
dB
0 10 20 30 40 50 60 70 80 90 100 110 120
FF l l
L/R
50
(%)
0
100
La Lettre d’ORL et de chirurgie cervico-faciale • n° 330 - juillet-août-septembre 2012 | 23
➤
fistule périlymphatique du fait de fluctuations auditives, en
cas de contexte traumatique ou barotraumatique ;
➤
de principe, une lésion évolutive, de type schwannome vesti-
bulaire, est évoquée, faisant réaliser une imagerie (IRM).
C’est l’association des 2 atteintes, oculaire et cochléo-vestibulaire,
et la récidive des signes auditifs à l’arrêt de la corticothérapie
qui permettent le diagnostic de syndrome de Cogan. L’avis du
médecin interniste est alors nécessaire, avec un double objectif,
diagnostique et thérapeutique.
Le bilan diagnostique recherche les autres atteintes possibles
au cours de cette maladie : articulaires, cardiaques avec insuffi-
sance aortique, vascularite, neurologique… Un amaigrissement
est parfois constaté.
Le traitement repose sur la corticothérapie au long cours et/ou
les immunosuppresseurs. Le médecin interniste décidera des
modalités : corticothérapie à la dose la plus faible possible, pour
laquelle les symptômes, en particulier auditifs, sont contrôlés, à
laquelle peuvent être ajoutés des immunosuppresseurs (métho-
trexate et azathioprine). Cette association thérapeutique permet,
dans un certain nombre de cas, de réduire la posologie des corti-
coïdes. La corticothérapie au long cours justifie des thérapeu-
tiques associées et une surveillance clinique et biologique (tension
artérielle, poids, ionogramme sanguin, glycémie, etc.), d’où l’intérêt
d’un suivi commun par le médecin interniste et l’ORL, afin de
vérifier l’absence d’effets indésirables et la stabilité de l’audition.
Dans certains cas, malgré ces mesures thérapeutiques, l’évolution
est marquée par une altération importante de l’audition, faisant
discuter une implantation cochléaire.

CAS CLINIQUE
Figure 2. Stratégie diagnostique d’une atteinte auditive potentiellement
auto-immune.
Surdité fluctuante
Surdité bilatérale rapidement évolutive
Symptomatologie de maladie de Ménière bilatérale
Et amélioration des symptômes durant la corticothérapie
Suspicion d’atteinte auto-immune
Évaluation clinique : atteinte oculaire, articulaire, état général…
Reprise de la corticothérapie
IRM éliminant une lésion évolutive, pathologie neurologique…
Avis de l’interniste : bilan diagnostique et thérapeutique adaptée
Surdités associées
à une maladie auto-immune
(syndrome de Cogan,
périartérite noueuse, etc.)
Surdités auto-immunes “isolées”
24 | La Lettre d’ORL et de chirurgie cervico-faciale • n° 330 - juillet-août-septembre 2012
En l’absence de tout symptôme oculaire, une surdité brusque
récidivante à l’arrêt des corticoïdes fera discuter une surdité auto-
immune. La description initiale des surdités auto-immunes a
été réalisée par B.F. Mc Cabe en 1979 (3, 4). Il s’agit de surdités
de perception bilatérales progressives sur plusieurs semaines ou
plusieurs mois, associées ou non à des troubles de l’équilibre,
pouvant se présenter comme une maladie de Ménière. La principale
caractéristique de ces surdités est l’efficacité de la corticothérapie,
avec parfois une corticodépendance : récidive des symptômes, en
particulier de la surdité, à l’arrêt des corticoïdes. Cette cortico-
dépendance peut conduire, là aussi, à proposer une corticothérapie
au long cours à des doses minimales, et/ou des thérapeutiques
immunosuppressives. En pratique, les situations cliniques pour
lesquelles la question d’une surdité auto-immune se pose sont les
surdités fluctuantes, les surdités bilatérales rapidement évolutives,
les patients présentant une maladie de Ménière bilatérale, en les
différenciant des surdités au cours d’affections auto-immunes
spécifiques (syndrome de Cogan, lupus érythémateux, périartérite
noueuse, etc.) [figure 2] (4).
Du point de vue physiopathologique, l’oreille interne peut être
le siège de réactions immunitaires au cours desquelles le sac
endolymphatique joue un rôle central. De nombreuses études
ont été réalisées pour identifier un test biologique spécifique
de ces surdités auto-immunes. Qu’il s’agisse de tests cellulaires
– tests d’inhibition de migration lymphocytaire ou de transfor-
mation lymphoblastique – ou de la recherche de certains anticorps
détectés par immunomarquage vis-à-vis de différentes protéines
– 68 (70) kDa, 30 kDa –, aucun de ces examens n’a démontré
un niveau de sensibilité et de spécificité le rendant utilisable en
pratique clinique. Le diagnostic est donc habituellement établi à
partir d’éléments biologiques non spécifiques : VS et CRP, et sur la
réponse à la corticothérapie, qui est donc un test diagnostique. Une
fois le diagnostic suspecté, la réalisation d’un bilan en médecine
interne permet d’éliminer une pathologie systémique dont la
surdité serait le premier symptôme et de fixer les modalités du
traitement et de son suivi. ■
1. Perdu J. Syndrome de Cogan : manifestations vasculaires et éléments du diagnostic.
Sang Thrombose Vaisseaux 2000;12(2):111-4.
2. Grasland A, Pouchot J, Hachulla E et al. Typical and atypical Cogan’s syndrome:
32 cases and review of the literature. Rheumatology (Oxford) 2004;43:1007-15.
3. McCabe BF. Autoimmune sensorineural hearing loss. Ann Otol Rhinol Laryngol
1979;88:585-9.
4. Vinceneux P, Couloigner V, Pouchot J, Bouccara D, Sterkers O. Surdités auto-
immunes. Presse Med 1999;28:1904-10.
Références bibliographiques
AVIS AUX LECTEURS
Les revues Edimark sont publiées en toute indépendance et sous l’unique et entière responsabilité du directeur de la publication et
du rédacteur en chef.
Le comité de rédaction est composé d’une dizaine de praticiens (chercheurs, hospi taliers, universitaires et libéraux), installés partout
en France, qui représentent, dans leur diversité (lieu et mode d’exercice, domaine de prédilection, âge, etc.), la pluralité de la disci-
pline. L’équipe se réunit 2 ou 3 fois par an pour débattre des sujets et des auteurs à publier.
La qualité des textes est garantie par la sollicitation systématique d’une relecture scientifique en double aveugle, l’implication d’un
service de rédaction/révision in situ et la validation des épreuves par les auteurs et les rédacteurs en chef.
Notre publication répond aux critères d’exigence de la presse :
• accréditation par la CPPAP (Commission paritaire des publications et agences de presse) réservée aux revues sur abonnements,
• adhésion au SPEPS (Syndicat de la presse et de l’édition des professions de santé),
• indexation dans la base de données INIST-CNRS, liens privilégiés avec la SFORL,
• déclaration publique de liens d’intérêts demandée à nos auteurs,
• identication claire et transparente des espaces publicitaires et des publi-rédactionnels en marge des articles scientiques.
1
/
3
100%