Dysgénésies gonadiques 46, XY et anomalies de la lignée

Cas clinique
mt Médecine de la Reproduction, Gynécologie Endocrinologie 2012 ; 14 (2) : 156-60
Dysgénésies gonadiques 46, XY et
anomalies de la lignée turnérienne
46, XY gonadal dysgenesis and features of Turner syndrome
Linda Pivois1
Sika Nassouri2
Franc¸oise Archambeaud2
Anne Drutel2
Sophie Galinat2
Stéphanie Lopez2
Marie-Pierre Teissier2
1Service d’Endocrinologie-Diabétologie,
CH Tulle, France
2Service de Médecine Interne
B- Endocrinologie,
Diabétologie,
Maladies métaboliques,
CHU Limoges,
87042 Limoges cedex, France
Résumé. Les dysgénésies gonadiques 46, XY sont révélées classiquement dans leur forme
pure par un impubérisme et une aménorrhée primaire chez une jeune fille, qui ne présente
par ailleurs aucune ambigüité sexuelle ni signe de virilisation, ni aucun stigmate évocateur
de syndrome de Turner. Une petite taille ou des anomalies phénotypiques caractéristiques
peuvent cependant être retrouvées, ce qui doit faire rechercher soit une mosaïque chromo-
somique comportant une lignée 45, X0, soit une délétion du bras court du chromosome Y.
Ces situations doivent conduire à un bilan complémentaire et à une surveillance, identiques
à ceux recommandés par la Haute Autorité de santé (HAS) dans le syndrome de Turner.
Mots clés : dysgénésie gonadique 46, XY, syndrome de Turner, mosaïque, gène SRY
Abstract. 46, XY complete gonadal dysgenesis often reveals as delayed puberty and primary
amenorrhea in a girl without ambiguous genitalia, virilization, or short stature. If present,
those last features can lead to detection of chromosomal mosaicism such as 45, X0/46, XY, or
deletion of the short arm of the Y chromosome. Initial and following evaluations must then be
performed as recommended in Turner syndrome.
Key words: 46, XY gonadal dysgenesis, Turner syndrome, mosaicism, SRY gene
Les dysgénésies gonadiques 46, XY
(DG 46, XY) sont des anomalies
de la détermination testiculaire en
rapport soit avec un défaut de signali-
sation par le gène SRY, soit avec une
anomalie d’un autre gène impliqué
dans la cascade de régulation déclen-
chée par SRY [1-3] (figure 1). Elles
se présentent selon plusieurs modali-
tés phénotypiques selon l’importance
des sécrétions testiculaires résiduelles
[1, 3]. Le phénotype externe et interne
peut en effet être masculin, ambigu
ou féminin sans ambiguïté sexuelle
comme dans la forme «pure », encore
appelée syndrome de Swyer, où les
gonades sont réduites à un stroma
indifférencié [1, 3]. Dans ce cas, les
DG 46, XY sont révélées par une
aménorrhée primaire associée à un
impubérisme, sans présence d’une
petite taille ou d’un syndrome dysmor-
phique comme dans le syndrome de
Turner (ST) [1, 3]. Des formes asso-
ciées existent également [1, 4, 5].
Observation
Nous rapportons le cas de Melle
B, dont le diagnostic de «dysgéné-
sie gonadique »a été évoqué à l’âge
de 13 ans devant un retard statu-
ral (-3,5 DS) et pubertaire avec un
appareil génital externe de type fémi-
nin. L’examen clinique retrouvait par
ailleurs un hypertélorisme mammaire,
une implantation basse des cheveux
et des oreilles. Les bilans biologiques
devaient révéler un hypogonadisme
périphérique, sans hypertestostéro-
némie, (l’hormone anti-müllerienne
n’ayant pas été dosée). Le caryo-
type sanguin standard est 46, XY
de formule homogène. L’exploration
cœlioscopique montrait un utérus et
des trompes d’aspect infantile, deux
gonades en bandelettes, qui ont été
réséquées. En histologie, ces dernières
étaient constituées d’un tissu fibreux
associé, pour la gonade droite, à
des structures testiculaires (cellules de
doi:10.1684/mte.2012.0401
médecine thérapeutique
Médecine
de la Reproduction
Gynécologie
Endocrinologie
Tirés à part : M.P. Teissier
156 Pour citer cet article : Pivois L, Nassouri S, Archambeaud F, Drutel A, Galinat S, Lopez S, Teissier MP. Dysgénésies gonadiques 46, XY et anomalies de la lignée
turnérienne. mt Médecine de la Reproduction, Gynécologie Endocrinologie 2012 ; 14 (2) : 156-60 doi:10.1684/mte.2012.0401
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 04/06/2017.

Leydig, tubes séminifères sans spermatogénèse) et
dénuées de toute lésion tumorale.
Le caryotype standard et la technique de fluorescence
in situ hybridization (FISH), effectués sur des cellules issues
de biopsies gonadiques et cutanées, ont permis d’isoler
une mosaïque 45, X0/46, XY. Le diagnostic de dysgénésie
gonadique mixte a donc été posé [4]. Elle a bénéficié après
la castration d’un traitement par hormone de croissance,
puis d’une supplémentation estro-progestative.
Au moment de sa prise en charge dans notre centre,
Mlle B est âgée de 18 ans. Elle est en bonne forme, mesure
1 m 60 et pèse 58 kg. Le contexte de DG, en mosaïque
Turner, nous a conduits à rechercher d’éventuelles
anomalies somatiques classiquement associées à ce syn-
drome [6]. Ce bilan s’est avéré normal en dehors de
la constatation de multiples nævi pour lesquels nous
avons préconisé des résections compte tenu du potentiel
carcinologique.
Discussion
Les dysgénésies gonadiques 46, XY sont classiquement
différenciées en formes «pures », sans activité gonadique
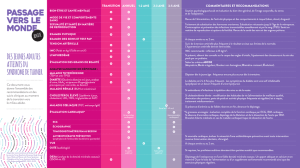
PAR 1
Bras court
(p)
SRY
Gène du
Iymphœdème
Gène du
gonadoblastome
Centromère
(c)
Bras long
(q)
Hétérochromatine
POF1
POF2
PAR2
Chromosome X
Shox
Shox
Centre
d’inactivation
Chromosome Y
Figure 1. Chromosomes X et Y. PAR = régions pseudo-autosomiques. SHOX = short stature homeobox-containing gene on the X-
chromosome. POF = primary ovarian failure.SRY=sex-determining region of Y chromosome.
masculine, et en formes «mixtes », où la persistance
d’îlots de tissu testiculaire peut être à l’origine d’une
sécrétion androgénique et donc de signes de virilisation
[1-3]. Notre observation est en cela atypique. En effet, le
tableau évoque une forme pure au plan clinique et bio-
logique, néanmoins l’analyse histologique des gonades
montre des cellules de Leydig et de Sertoli, la classant en
“mixte”. Toutefois, le retard pubertaire, le phénotype pure-
ment féminin et la testostéronémie effondrée, ainsi que
l’absence de résidus wolffiens en cœlioscopie, font penser
que les cellules de Leydig isolées dans la bandelette sont
non fonctionnelles. De même, nous formulons l’hypothèse
que les cellules de Sertoli, également présentes, n’étaient
pas opérantes compte tenu de l’absence de spermatoge-
nèse et de la persistance chez notre patiente des structures
müllériennes (utérus et trompes), ce qui signe l’absence
d’hormone anti-müllérienne (AMH), mais malheureuse-
ment les dosages n’ont pas été effectués [1-3]. Par ailleurs,
le tableau phénotypique de Mlle B s’associe à un retard
de croissance et des éléments dysmorphiques très évoca-
teurs d’un ST en dépit d’un caryotype sanguin de formule
46, XY homogène. Actuellement, la FISH est réalisée
de fac¸on systématique sur lymphocytes en complément
du caryotype standard pour rechercher les mosaïques
mt Médecine de la Reproduction, Gynécologie Endocrinologie, vol. 14, n◦2, avril-mai-juin 2012 157
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 04/06/2017.

Cas clinique
Tableau 1. Recommandations de surveillance des patientes atteintes de syndrome de Turner [6].
Au diagnostic Enfance Age adulte
Examen clinique détaillé :
poids, taille, TA, recherche de
strabisme, cyphose, scoliose
Oui Tous les ans Tous les ans
Glycémie à jeun
±HbA1c
±HGPO
Après 10 ans Tous les 2 ans (après 10 ans) ou
avant traitement par GH
Tous les ans
ASAT, ALAT, ␥GT, PAL Après 6 ans Tous les 2 ans Tous les ans
Créatininémie Si malformation rénale Si malformation rénale Si malformation rénale ou HTA
Bilan lipidique Après 10 ans Tous les 2 ans (après 10 ans) Tous les ans
TSH ±T4L
Ac anti-TPO
Après 4 ans Tous les 2 ans si Ac anti-TPO
négatifs
Tous les ans si Ac anti-TPO
positifs
Tousles1à2anssiAcanti-TPO
négatifs
Tous les ans si Ac anti-TPO positifs
FSH ±LH Oui Contrôle avant induction
pubertaire
Ac anti-transglutaminase (IgA) Après 4 ans Tous les 2 ans Tous les 2 ans
IGF1 Début traitement GH
Tous les 6-12 mois si traitement
GH
Consultation cardiologique
+ échographie cardiaque
Oui Tous les ans si pathologie
cardiaque et/ou HTA
Tous les 5 ans si absence de
cardiopathie et d’HTA
Tous les ans si pathologie cardiaque
et/ou HTA
Tous les 5 ans si absence de
cardiopathie et d’HTA
IRM aortique Selon avis cardiologique Selon avis cardiologique Selon avis cardiologique
Échographie rénale Oui
Échographie pelvienne Avant induction pubertaire
Contrôle en fin de croissance
ou puberté
Avant induction pubertaire
Contrôle en fin de croissance
ou puberté
Préparation utérine hormonale ou
suivi de grossesse
Échographie thyroïdienne Si dysthyroïdie, nodule palpé
ou goitre
Si dysthyroïdie, nodule palpé
ou goitre
Si dysthyroïdie, nodule palpé ou
goitre
Âge osseux Oui Tous les1à3anssous GH
Ostéo-densitométrie Avant induction pubertaire
Contrôle en fin de croissance
ou puberté
Avant induction pubertaire
Contrôle en fin de croissance
ou puberté
Tous les 5 ans
Consultation ORL et étude
audition
Oui Tous les 2 ou 3 ans minimum
ou plus rapproché selon avis
ORL
Tous les 2 ou 3 ans minimum ou
plus rapproché selon avis ORL
Consultation ophtalmologie Oui Selon avis ophtalmologique Selon symptômes
Consultation stomatologie Après 6 ans (avant, si anomalie
clinique)
Selon clinique Selon clinique
Consultation diététique Si surpoids, intolérance au
glucose, diabète, dyslipidémie
Si surpoids, intolérance au
glucose, diabète, dyslipidémie
Si surpoids, intolérance au glucose,
diabète, dyslipidémie
Consultation psychologue ±
tests psychométriques
Oui Vers l’âge de 4 ou 5 ans (avant
si signes d’appel)
Réévaluation selon symptômes
Selon symptômes
Coloscopie Proposée à partir de l’âge de 45 ans
puis tous les 5 ans
TA = tension artérielle. HGPO = hyperglycémie provoquée orale. GH = growth hormone (hormone de croissance). ASAT = aspartate ami-
notransférase. ALAT = alanine aminotransférase. ␥GT = gammaglutamyltranspeptidase. PAL = phosphatases alcalines. HTA = hypertension
artérielle. TSH = thyroid stimulating hormone. Ac = anticorps. TPO = thyroperoxydase. FSH = follicle-stimulating hormone. LH = luteinizing
hormone. IgA = immunoglobuline A. IGF1 = insulin growth factor 1.
158 mt Médecine de la Reproduction, Gynécologie Endocrinologie, vol. 14, n◦2, avril-mai-juin 2012
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 04/06/2017.

gonosomiques en cas de 46XY avec phénotype fémi-
nin comme en cas de 45X0 [1, 4]. Le contexte justifie
de poursuivre la recherche diagnostique d’éléments de
certitude de la présence ou non d’un contingent cellu-
laire turnérien. L’identification d’une mosaïque tissulaire
(cutanée et gonadique) de type 45, X0/46, XY justifie
d’un traitement adapté dans l’enfance et de conduire
le bilan d’éventuelles malformations et/ou patholo-
gies, classiquement associées au ST (tableau 1, d’après
l’HAS).
La revue de la littérature souligne que les modalités
de suivi des patientes présentant une DG «associée »
sous la forme d’une mosaïque tissulaire sont imprécises.
Notre propos ne concerne pas les cas de mosaïques 45,
X0/46, XY évidentes sur le caryotype sanguin, puisqu’elles
renvoient à la prise en charge d’un ST avec présence de
chromosome Y (5 % des cas de syndrome de Turner)
[7-9]. En effet, il est établi que ces anomalies caryo-
typiques lymphocytaires en mosaïque sont volontiers
associées à des malformations, notamment cardiovas-
culaires [1, 9]. Le bilan et la surveillance sont donc
ceux du ST avec nécessité d’une gonadectomie bilatérale
prophylactique, en raison des risques de virilisation et sur-
tout de dégénérescence de ces gonades dysgénésiques Y
[6-9].
Notre observation souligne la nécessité d’évoquer et
de rechercher une dysgénésie gonadique associée chez la
fille présentant une dysgénésie gonadique 46, XY homo-
gène sur le prélèvement sanguin, en présence d’anomalies
cliniques turnériennes. Cette association pourrait renvoyer
à deux entités génétiques différentes : soit une mosaïque
chromosomique «occulte »45, X0/46, XY, soit une
délétion du bras court du chromosome Y (Yp) qui corres-
pond aux dysgénésies gonadiques 46, XY SRY-négatives
[1, 4, 5, 10]. La fréquence de ces deux situations au
sein des dysgénésies 46, XY n’est pas précisément connue
[1-3]. Nous savons cependant que les anomalies du gène
SRY sont rencontrées dans 10 à 20 % des cas de dysgé-
nésie gonadique 46, XY, et correspondent essentiellement
à des délétions de la région SRY après translocation des
régions pseudo-autosomiques des chromosomes sexuels
au cours de la méiose paternelle [1-3]. Les descriptions
de mosaïques tissulaires 45, X0/46, XY avec formule san-
guine 46, XY, comme notre patiente, sont quant à elles
exceptionnelles [4, 5]. Les modalités de suivi sont tout
aussi imprécises.
Il apparaît donc nécessaire, dans la prise en charge
des dysgénésies gonadiques 46, XY, d’orienter l’examen
clinique vers la recherche de signes évocateurs de ST.
En présence de telles anomalies cliniques, le bilan doit
absolument rechercher une mosaïque 45, X0/46, XY
tissulaire ou une délétion du bras court du chromosome Y
[1]. Rappelons qu’en pratique, la recherche de mosaïque
repose sur le caryotype standard sur 30 cellules, souvent
complété par une étude de cytogénétique utilisant la
technique de FISH qui permet d’analyser un nombre plus
important de noyaux cellulaires [6, 7]. Ces explorations
cytogénétiques peuvent être réalisées sur prélèvement
sanguin, mais aussi sur frottis buccal, voire sur une biopsie
cutanée ou gonadique comme dans le cas de Mlle B.
[1, 4-6, 9]. D’autre part, pour objectiver une délétion du
bras court du chromosome Y, sont utilisées généralement
des sondes spécifiques du locus SRY, soit en cytogénétique
(FISH), soit en biologie moléculaire par polymerase chain
reaction (PCR) [1, 4-6, 9] (figure 1). La mise en évidence
de cellules 45, X0 ou 46, XY délétées en Yp doit conduire
à une surveillance identique à celle du ST et, à l’âge
pédiatrique, à un traitement par hormone de croissance
[1]. Dans notre observation, le bilan initial systématique à
la recherche de malformations associées au ST s’est avéré
négatif, à l’exception de la constatation de nævi pigmen-
taires multiples. Son suivi à 4 ans révèle d’autres naevi à
réséquer et l’apparition d’un surpoids androïde associé à
une dyslipidémie et à une HTA qui peuvent être rapportés
au ST. La normalité des premiers examens ne doit pas
faire omettre la surveillance ultérieure recommandée par
les autorités de santé pour le ST [6].
Conclusion
Tout comme il est recommandé de chercher du chro-
mosome Y dans le syndrome de Turner 45, X0 [6, 7, 9],
nous pensons qu’il faut évoquer la présence d’une «mono-
somie X »dans les dysgénésies gonadiques 46, XY chez
la fille. Ceci renvoie aux mosaïques 45, X0/46, XY, ainsi
qu’aux délétions du bras court du chromosome Y (dysgé-
nésies gonadiques 46, XY SRY-négative) qui conduisent
aux mêmes situations d’haplo-insuffisance des gènes de
l’X. Cette notion cytogénétique devra orienter l’examen
clinique à la recherche de petits signes de la lignée
turnérienne, et amener en leur présence à approfon-
dir les explorations génétiques. Le rationnel de cette
démarche est la nécessité de pratiquer chez ces patientes
des bilans réguliers à la recherche de malformations
et anomalies classiquement associées au syndrome de
Turner.
Références
1. Ostrer H. 46, XY disorder of sex development and 46, XY complete
gonadal dysgenesis. In : Pagon RA, Bird TD, Dolan CR, Stephens K,
eds. GeneReviews. Seattle (WA) : University of Washington, Seattle ;
1993. http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book¼gene
& part¼gonad-dys-46xy.
2. Coutin AS, Hamy A, Fondevilla M, Savigny B, Paineau J, Visset J. La
dysgénésie gonadique pure à 46, XY. J Gynecol Obstet Biol Reprod
1996 ; 25 : 792-6.
mt Médecine de la Reproduction, Gynécologie Endocrinologie, vol. 14, n◦2, avril-mai-juin 2012 159
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 04/06/2017.

Cas clinique
3. Kuttenn F, d’Acremont MF, Mowszowicz I. Anomalies de la dif-
férenciation sexuelle. Encycl Med Chir, Endocrinologie-Nutrition,
10-033-A-10, 2003, 26 p.
4. Röpke A, Pelz AF, Volleth M, Schlösser HW, Morlot S, Wieacker
PF. Sex chromosomal mosaicism in the gonads of patients with gona-
dal dysgenesis, but normal female or male karyotypes in lymphocytes.
Am J Obstet Gynecol 2004 ; 190 : 1059-62.
5. Röpke A, Kalinski T, Mohnike K, et al. Distribution of sex chromo-
somes in dysgenetic gonads of mixed type. Cytogenet Genome Res
2007 ; 116 : 146-51.
6. Anonyme. Syndrome de Turner. Protocole ALD n◦31. Protocole
National de Diagnostic et de Soins (PNDS). Guides ALD. Jan-
vier 2008.
7. Cabrol S. Syndrome de Turner. Encyclopédie Orphanet.
Février 2007. Available from : URL : http://www.orpha.net/data/
patho/Pro/fr/Turner-FRfrPro44.pdf.
8. Cormier-Daire V, Bouvattier C. Syndrome de Turner : corrélations
entre phénotypes et défauts cytogénétiques du chromosome X. In :
Pienkowski C, Tauber M. Le syndrome de Turner. Springer-Verlag
France ; 2009 : 37-54.
9. Sybert VP, McCauley E. Turner’s syndrome. N Engl J Med
2004 ; 351 : 1227-38.
10. Boucher CA, Sargent CA, Ogata T, Affara NA. Breakpoint
analysis of Turner patients with partial Xp deletions : implica-
tions for the lymphoedema gene location. J Med Genet 2001;38:
591-8.
160 mt Médecine de la Reproduction, Gynécologie Endocrinologie, vol. 14, n◦2, avril-mai-juin 2012
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 04/06/2017.
1
/
5
100%