1. DÉNOMINATION DU MÉDICAMENT Paracetamol AB 10 mg/ml

1. DÉNOMINATION DU MÉDICAMENT
Paracetamol AB 10 mg/ml solution pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Un millilitre contient 10 mg de paracétamol.
Un flacon de 50 ml contient 500 mg de paracétamol.
Un flacon de 100 ml contient 1000 mg de paracétamol.
Excipients : sodium 0,076 mg/ml.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Solution pour perfusion.
La solution est limpide, légèrement jaunâtre et dépourvue de particules.
pH 5,5
Osmolarité 295 mOsm/litre
4. DONNÉES CLINIQUES
4.1 Indications thérapeutiques
Paracetamol AB est indiqué pour le traitement à court terme des douleurs modérées, en particulier
après une intervention chirurgicale et le traitement à court terme de la fièvre,
lorsque l’administration par voie intraveineuse est cliniquement justifiée par une nécessité urgente
de traiter la douleur ou l’hyperthermie et/ou lorsque d’autres voies d’administration sont
impossibles.
4.2 Posologie et mode d’administration
Voie intraveineuse.
Le flacon de 50 ml est réservé aux nouveau-nés à terme, aux nourrissons, aux jeunes enfants et aux
enfants pesant moins de 33 kg.
Le flacon de 100 ml est réservé aux adultes, aux adolescents et aux enfants pesant plus de 33 kg.
Posologie :
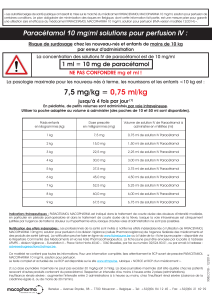
Posologie en fonction du poids du patient (veuillez consulter le tableau de posologie ci-dessous)
1/10

Poids du patient Dose
(par administration)
Volume par
administration
Volume maximal
de paracetamol,
solution pour
perfusion
(10 mg/ml) par
administration,
sur base des
limites
supérieures de
poids du groupe
(ml)***
Dose quotidienne
maximale **
< 10 kg * 7,5 mg/kg 0,75 ml/kg 7,5 ml 30 mg/kg
>10 kg et ≤ 33 kg 15 mg/kg 1,5 ml/kg 49,5 ml 60 mg/kg, sans
dépasser 2 g
> 33 kg et
≤ 50 kg
15 mg/kg 1,5 ml/kg 75 ml 60 mg/kg, sans
dépasser 3 g
> 50 kg, avec des
facteurs de
risque
supplémentaires
d’hépatotoxicité
1 g 100 ml 100 ml 3 g
> 50 kg, sans
facteurs de
risque
supplémentaires
d’hépatotoxicité
1 g 100 ml 100 ml 4 g
* Nouveau-nés prématurés : On ne dispose d’aucune donnée de sécurité et d’efficacité pour les
nouveau-nés prématurés (voir rubrique 5.2).
**Dose quotidienne maximale : La dose quotidienne maximale présentée dans le tableau ci-dessus
s’applique aux patients ne recevant aucun autre produit à base de paracétamol et cette dose doit
être ajustée en tenant compte de la prise éventuelle de ces produits.
***Les patients de poids inférieur nécessiteront des volumes plus faibles.
L’intervalle minimal entre chaque administration doit être d’au moins 4 heures.
Chez les patients ayant une insuffisance rénale sévère, l’intervalle minimal entre chaque
administration doit être d’au moins 6 heures.
Ne pas administrer plus de 4 doses en 24 heures
Insuffisance rénale sévère : lorsqu’on administre du paracétamol à des patients souffrant
d'insuffisance rénale sévère (clairance de la créatinine ≤ 30 ml/min), il est recommandé de réduire la
dose et d’allonger l'intervalle minimal entre chaque administration à 6 heures (voir rubrique 5.2).
Chez les adultes souffrant d’insuffisance hépatocellulaire, d’alcoolisme chronique, de malnutrition
chronique (réserves basses en glutathion hépatique), de déshydratation :
La dose journalière maximale ne doit pas dépasser 3000 mg (voir rubrique 4.4).
2/10

Mode d’administration :
Lorsqu’on prescrit et administre paracetamol solution pour perfusion, veiller à éviter les erreurs de
dosage en raison d’une confusion entre les milligrammes (mg) et les millilitres (ml), qui pourraient
donner lieu à un surdosage accidentel et au décès. S’assurer que la dose correcte a bien été
communiquée et délivrée. Lorsqu’on rédige les prescriptions, indiquer la dose totale en mg et la dose
totale en volume. S’assurer que la dose est mesurée et administrée avec précision.
La solution de paracétamol est administrée au moyen d’une perfusion intraveineuse de 15 minutes.
Patients ayant un poids ≤ 10 kg :
•Le flacon en verre/la poche de Paracetamol AB ne doit pas être installé(e) sur un pied à
perfusion, vu le faible volume de médicament à administrer dans cette population.
•Le volume à administrer doit être prélevé du flacon et dilué dans une solution de chlorure de
sodium à 0,9 % ou de glucose à 5 % jusqu’à l’obtention d’une dilution maximale d’un dixième (un
volume de Paracetamol AB dans neuf volumes de diluant), puis administré sur une durée de 15
minutes.
•Utiliser une seringue de 5 ou 10 ml pour mesurer la dose correcte en fonction du poids de
l’enfant et le volume souhaité, qui ne doit néanmoins jamais dépasser 7,5 ml par dose.
•Inviter l’utilisateur à consulter les informations de la notice relatives à la posologie.
Pour prélever la solution, utiliser une aiguille de 0,8 mm (21 G) et perforer le bouchon à la verticale à
l’endroit précisé.
Flacon de 50 ml :
Le contenu du flacon de 50 ml de Paracetamol AB peut également être dilué dans une solution de
chlorure de sodium à 0,9 % ou de glucose à 5 % jusqu’à l’obtention d’une dilution maximale d’un
dixième (un volume de Paracetamol AB dans neuf volumes de diluant). Dans ce cas, utiliser la
solution diluée dans les quatre heures suivant sa préparation (temps de perfusion inclus).
Comme pour toutes les solutions pour perfusion conditionnées dans des flacons en verre, il est
rappelé qu’une surveillance étroite est nécessaire, particulièrement à la fin de la perfusion, quelle
que soit la voie d’administration. Cette surveillance à la fin de la perfusion s’applique tout
particulièrement aux perfusions administrées par voie centrale, de façon à éviter une embolie
gazeuse.
4.3 Contre-indications
Paracetamol AB est contre-indiqué :
-chez les patients présentant une hypersensibilité au paracétamol ou au chlorhydrate de
propacétamol (prodrogue du paracétamol) ou à l'un des excipients mentionnés à la rubrique 6.1
-en cas d’insuffisance hépatocellulaire sévère.
4.4 Mises en garde spéciales et précautions d’emploi
3/10

Mises en garde
RISQUE D’ERREURS DE DOSAGE
Veiller à éviter les erreurs de dosage secondaires à une confusion entre les milligrammes (mg) et
les millilitres (ml), qui pourraient donner lieu à un surdosage accidentel et au décès (voir rubrique
4.2).
Il est recommandé d’utiliser un traitement antalgique oral adapté dès que cette voie
d'administration est possible.
Pour éviter le risque de surdosage, il faut vérifier l'absence de paracétamol ou de propacétamol dans
la composition d'autres médicaments associés.
Des doses supérieures aux doses recommandées entraînent un risque d'atteinte hépatique très
sévère. Les signes cliniques et les symptômes d'atteinte hépatique (y compris hépatite fulminante,
insuffisance hépatique, hépatite cholestatique, hépatite cytolytique) ne s’observent habituellement
pas avant deux jours, et jusqu’à un maximum de 4-6 jours après l’administration. Le traitement par
antidote doit être initié le plus rapidement possible (voir rubrique 4.9).
Ce médicament contient moins de 1 mmol de sodium (23 mg) par 100 ml, il est donc essentiellement
« sans sodium ».
Précautions d’emploi
Le paracétamol doit être utilisé avec précaution en cas :
-d'insuffisance hépatocellulaire
-d'insuffisance rénale sévère (clairance de la créatinine ≤ 30 ml/min) (voir rubriques 4.2 et 5.2)
-d'alcoolisme chronique
-de malnutrition chronique (réserves faibles en glutathion hépatique)
-de déshydratation.
4.5 Interactions avec d’autres médicaments et autres formes d’interactions
-Le probénécide réduit environ de moitié la clairance du paracétamol en bloquant sa conjugaison
avec l'acide glucuronique. Une réduction de la dose de paracétamol doit être envisagée lors d'un
traitement simultané avec du probénécide.
-La salicylamide peut prolonger la demi-vie d'élimination du paracétamol.
-La vigilance est de rigueur lors de l'administration simultanée de substances inductrices
d'enzymes (voir rubrique 4.9).
- L’utilisation concomitante de paracétamol (4 g par jour pendant au moins 4 jours) et
d’anticoagulants oraux peut entraîner de légères variations des valeurs d’INR. Dans ce cas, une
surveillance accrue des valeurs d’INR est nécessaire pendant la période d’utilisation
concomitante et 1 semaine après l’arrêt du traitement par paracétamol.
4.6 Fertilité, grossesse et allaitement
Grossesse :
L'expérience clinique de l'administration intraveineuse de paracétamol est limitée. Toutefois, les
données épidémiologiques disponibles sur l’utilisation de doses thérapeutiques de paracétamol par
voie orale n'indiquent aucun effet indésirable sur la grossesse ou la santé du fœtus / nouveau-né.
Des données prospectives concernant les grossesses exposées à des surdosages n’ont pas montré de
risque accru de malformations.
4/10

On n’a pas conduit d’études de reproduction animale avec la forme intraveineuse de paracétamol.
Toutefois, les études effectuées avec la forme orale n'ont pas démontré de malformations ni d’effets
fœtotoxiques.
Néanmoins, Paracetamol AB ne doit être utilisé pendant la grossesse qu'après une évaluation
soigneuse du rapport bénéfices/risques. Dans ce cas, la posologie et la durée de traitement
recommandées doivent être observées strictement.
Allaitement :
Après une administration orale, le paracétamol est excrété en petites quantités dans le lait maternel.
Aucun effet indésirable n'a été rapporté chez les enfants allaités. Par conséquent, Paracetamol AB
peut être utilisé par des femmes qui allaitent.
4.7 Effets sur l’aptitude à conduire des véhicules et à utiliser des machines
Le paracétamol n’a aucune influence sur l’aptitude à conduire des véhicules et à utiliser des
machines.
4.8 Effets indésirables
Au sein de chaque groupe de fréquence, les effets indésirables doivent être présentés suivant un
ordre décroissant de gravité.
Comme c’est le cas avec tous les produits contenant du paracétamol, les effets indésirables sont
rares (≥1/10000 à <1/1000) ou très rares (<1/10000). Ils sont décrits ci-dessous :
Systèmes d’organes Rare
≥1/10000 à <1/1000
Très rare
(<1/10000)
Affections hématologiques et
du système lymphatique
Thrombocytopénie, Leucopénie,
Neutropénie.
Affections cardiaques Hypotension
Affections hépatobiliaires Élévation des taux des
transaminases hépatiques
Troubles généraux et
anomalies au site
d’administration
Malaise Réaction d’hypersensibilité
Des réactions indésirables fréquentes au site d'injection ont été rapportées pendant les études
cliniques (douleurs et sensation de brûlure).
De très rares cas de réactions d’hypersensibilité, allant de simples éruptions cutanées ou d’urticaire
au choc anaphylactique ont été rapportés et nécessitent l'arrêt du traitement.
Des cas d'érythème, de bouffées vasomotrices, prurite et de tachycardie ont été rapportés.
De très rares cas de réactions cutanées sévères ont été rapportés.
Déclaration des effets indésirables suspectés
5/10
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%