Certificat d`enregistr

APPROUVÉ
Ordre du Ministère de la protection
de la santé de l'Ukraine
_____________ № ______________
Certificat d'enregistrement
№ _____________________
FICHE POSOLOGIQUE d’un médicament
INFULGAN®
Composition :
Principe actif: paracétamol;
1 ml de solution contient 10 mg de paracétamol;
Excipients : acide citrique, monohydrate; citrate de sodium; sorbite (Е 420), sodium
sulfite anhydre (Е 221), eau pour injection.
Forme pharmaceutique. Solution pour perfusion.
Groupe pharmacothérapeutique.
Analgésiques et antipyrétiques. Code ATC N02B E01.
Données cliniques.
Indications.
Adultes : traitement à court terme du syndrome douloureux d'intensité moyenne,
surtout dans la période post-opératoire.
Traitement à court terme des réactions hyperthermiques.
Enfants: traitement symptomatique de la douleur et de l'hyperthermie dans la période
post-opératoire.
Contre-indications.
Hypersensibilité au paracétamol et autres constituants du produit. Insuffisance
hépatocellulaire sévère.
Posologie et mode d’emploi.
Infulgan est destiné au soulagement rapide du syndrome douloureux et/ou
hyperthermique, quand l’injection d’un médicament seulement par voie intraveineuse
est nécessaire.
En cas d'administration de la préparation aux enfants, avant la perfusion il faut retirer
du flacon l’excédent du médicament et laisser le volume de la solution correspondant à
une dose.
La perfusion intraveineuse doit durer 15 minutes.
Adultes et enfants avec un poids corporel de 50 kg ou plus.
La dose maximale par prise est de 1000 mg de paracétamol.
La dose maximale par jour est de 4 g. L'intervalle entre les injections du médicament
ne doit pas être moins de 4 heures. Généralement de 1 à 4 perfusions seront
administrées durant les premierès 24 heures dès l’apparition du syndrome douloureux
(période post-opératoire), si nécessaire, la durée du traitement peut être prolongée,
cependant elle ne doit pas dépasser 72 heures, le nombre total de perfusions ne doit pas
excéder 12 fois.
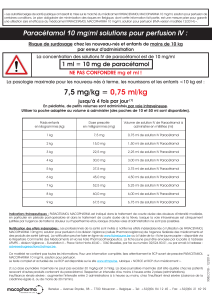
Enfants avec un poids corporel de 33 kg à 50 kg.
15 mg/kg de paracétamol par perfusion, c'est-à-dire 1,5 ml/kg. La dose maximale par
jour ne doit pas dépasser 60mg/kg de poids corporel. L'intervalle minimal entre les
injections est de 4 heures. La durée de traitement ne dépasse pas généralement 4
perfusions pendant 24 heures.
Enfants avec un poids corporel de 10 kg à 33 kg.

15 mg/kg de paracétamol par injection, c'est-à-dire 1,5 ml/kg. La dose maximale par
jour ne doit pas dépasser 60mg/kg de poids corporel. L'intervalle minimal entre les
injections est de 4 heures. La durée du traitement ne dépasse pas généralement 4
perfusions pendant 24 heures.
Chez les adultes atteints de l'insuffisance rénale (clairance de la créatinine 30
ml/min) l'intervalle entre les prises doit augmenter jusqu'à 6 heures. La durée du
traitement ne doit pas dépasser 48 heures.
Effets indésirables.
Systèmes
d’organes
Effets rares
>1/10000,
<1/1000
Effets très
rares
<1/10000
Rapports
particuliers
Réactions
générales
Malaise
Réactions
de
l'hypersensi
bilité
Choc
anaphylactique
Système
cardiovasculaire
Hypotension
artérielle
Appareil digestif
Augmentation
du taux de
transaminases
hépatiques
Sang
Thrombocytopé
nie, leucopénie,
neutropénie
Dans les cas particuliers des éruptions cutanées ordinaires ou urticariennes étaient
observées, les cas de choc anaphylactique et de thrombocytopénie sont également
connus.
Surdosage.
Le risque d'action toxique du produit est élevé chez les personnes âgées, les enfants,
les patients atteints de l'insuffisance hépatique, en cas de l'alcoolisme chronique, de
dystrophie alimentaire et chez les personnes atteintes de l'activité enzymatique réduite.
Dans les cas indiqués ci-dessus le surdosage peut être mortel.
Les symptômes apparaissent pendant les premières 24 heures et se manifestent par les
nausées, les vomissements, l'anorexie, la pâleur, les douleurs abdominales.
Le surdosage chez les adultes peut avoir lieu en cas d’injection en une seule fois d’une
dose de 7,5 g ou plus, chez les enfants - 140 mg/kg de poids corporel. Dans ces cas il se
développe la cytolyse hépatique, l'insuffisance hépatique, l'acidose métabolique,
l'encéphalopathie pouvant causer le coma et la mort du patient. Pendant 12-48 heures le
taux de transaminases hépatiques (ASAT, ALAT), de lactate-déhydrogénase, de
bilirubine augmente et le taux de prothrombine diminue. Les symptômes cliniques
d’affection du foie se manifestent après deux jours et atteignent leur maximum après 4-
6 jours.
Traitement : introduction des donateurs des groupes sulfhydryles et des précurseurs de
synthèse de glutathion-méthionine dans 8-9 heures après le surdosage et de N-
acétylcystéine - dans 12 heures. La nécessité de la tenue des actions thérapeutiques
complémentaires (introduction ultérieure de méthionine, injection intraveineuse de N-
acétylcystéine) doit être définie en fonction de la concentration de paracétamol dans le
sang, et également du temps passé après sa prise.
Grossesse et allaitement.

Il n’y a pas de données concernant l'influence négative de l'administration intraveineuse
de paracétamol sur le développement du fœtus ou les effets fetotoxiques, cependant
avant l'utilisation du médicament il faut évaluer attentivement le rapport profit/risque,
pendant l'administration du produit la femme enceinte doit être soigneusement suivie.
Le paracétamol est excrété dans le lait maternel. Pendant le traitement par le produit il
est nécessaire d’arrêter l’allaitement.
Enfants.
Le produit peut être administré aux enfants à l'âge de 1 an de poids corporel de plus de
10 kg seulement pour le traitement symptomatique de la douleur et de l'hyperthermie
chez les sujets dans la période post-opératoire.
Précautions particulières.
Le produit doit être administrée avec prudence chez les patients souffrant de :
- insuffisance hépatocellulaire;
- insuffisance rénale sévère (clairance de la créatinine moins de 30 ml/min);
- alcoolisme chronique;
- carence alimentaire (diminution de la réserve en glutathion dans le foie);
- déshydratation.
Le risque du développement d’affections du foie pendant le traitement par Infulgan est
élevé chez les malades atteints de l’hépatose alcoolique.
L'administration d'Infulgan peut influencer négativement les résultats des analyses de
laboratoire lors de la détermination quantitative du contenu du glucose et l'acide urique
dans le plasma sanguin.
Pendant le traitement à long terme le contrôle au niveau du sang périphérique et de l'état
fonctionnel du foie est nécessaire.
Le médicament doit être utilisé une seule fois, le reste ne doit pas être utilisé. Ne pas
utiliser après la date d’échéance figurant sur l'emballage.
Effets sur l’aptitude à conduire des véhicules et à utiliser des machines.
Le produit n'a aucune influence sur la conduite de véhicules ou l'utilisation de
machines.
Interactions avec d'autres médicaments et d'autres formes d’interactions.
Le probénécide réduit doublement la clairance de paracétamol par voie de blocage de
sa conjugaison à l'acide glucuronique, c'est pourquoi lors de la thérapie associée avec
l’utilisation de probénécide il est nécessaire de réduire la dose du paracétamol.
Les salicylates peuvent augmenter la demi-vie d’élimination du paracétamol de
l'organisme.
Les inducteurs de l’oxydation microsomale dans le foie (phénytoïne, alcool éthylique,
barbituriques, rifampicine , phénylbutazone, antidépresseurs tricycliques) peuvent
contribuer au développement des intoxications sévères même en cas d’un petit
surdosage.
Propriétés pharmacologiques.
Pharmacodynamie. L’infulgan (paracétamol) produit un effet analgésique et fébrifuge.
Le paracétamol bloque la cyclo-oxygénase (COX) I et II uniquement dans le système
cérébro-spinal, en influençant les centres de la douleur et de la thermorégulation. Les
peroxydases cellulaires neutralisent l'effet du paracétamol sur COX dans les tissus
excités, ce qui explique l'absence presque complète de l'effet antiinflammatoire.
L'absence d'effet sur la synthèse des prostaglandines dans les tissus périphériques
conditionne l'absence d’impact négatif sur les échanges hydro-salins (rétention de
sodium et rétention hydrique) et sur la muqueuse du tube digestif.

Pharmacocinétique. La concentration maximale dans le plasma sanguin serait atteinte
dans 15 minutes; la concentration maximale est de 15-30 mkg/ml. Le volume de
distribution est de 1 l/kg. Le paracétamol est faiblement lié aux protéines plasmatiques;
il pénètre par la barrière hémato-encéphalique; il est métabolisé dans le foie avec la
formation des glucuronides et des sulfates. Une petite partie (4 %) est métabolisée par le
cytochrome Р450 avec la formation intermédiaire de métabolite (N -acetyl-
benzochinonimine), ce qui dans les conditions normales d’utilisation est rapidement
détoxifié par le glutathion réduit et éliminé dans les urines après la conjugaison à la
cystéine et à l'acide mercaptopurique. Cependant en cas d'intoxication massive la
quantité de ce métabolite toxique augmente. La demi-vie d’élimination chez les adultes
est de 2,7 heures, chez les enfants est de 1,5-2 heures, chez les bébés est de 3,5 heures,
la clairance totale est de 18 l/heure. Le paracétamol est éliminé essentiellement dans les
urines; 90 % de la dose prise sont éliminés par les reins pendant 24 heures,
principalement sous la forme de glucuronide (60-80 %) et de sulfate (20-30 %). Moins
de 5 % sont éliminés dans un état inchangé. En cas d'insuffisance rénale sévère
(clairance de la créatinine est inférieure à 10-30 ml/min) l’élimination de paracétamol
est un peu ralenti, et la demi-vie d’élimination est de 2-5,3 heures. La vitesse
d’élimination du glucuronide et du sulfate chez les patients atteints de l'insuffisance
rénale sévère est 3 fois moins que chez les volontaires en bonne santé.
La pharmacocinétique chez les enfants ne se distingue pas pratiquement d'une celle des
adultes, à l'exception de la demi-vie d’élimination plus courte du plasma sanguin (1,5-2
heures). Chez les enfants de moins de 10 ans la conjugaison à l'acide glucuronique est
considérablement réduite, et la conjugaison aux sulfates est plus augmentée par rapport
aux adultes.
Caractéristiques pharmaceutiques.
Principales propriétés physico-chimiques: solution transparente, incolore ou quelque
peu jaunâtre.
Validité.
2 ans. Ne pas utiliser après la date d’échéance figurant sur l'emballage.
Conservation.
Ne pas laisser à la portée des enfants.
Conserver à l'abri de la lumière et à une température ne dépassant pas 25° C. Ne pas
congeler.
Conditionnements. Bouteilles de 20 ml, 50 ml ou 100 ml. Boîtes d’une bouteille.
Mode de délivrance.
Sur prescription médicale.
Fabricant.
SARL « YURIA-PHARM ».
Siège.
SARL « YURIA-PHARM », r. M. Amosova, 10, Kiev, 03680, Ukraine
Tél./fax (044) 275-92-42; 275-01-08.
Date de la dernière modification.
1
/
4
100%