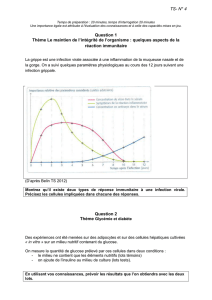
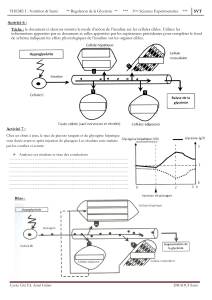
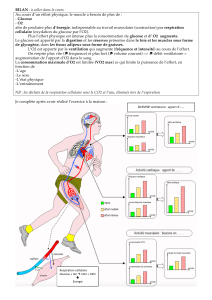
Homéostasie

Homeostasie et Nutrition
Ecole d’été
Département Alimentation Humaine
6 au 9 juillet 2009


C
’est au cœur de la Normandie, dans le village d’Etretat que le
Département Alimentation Humaine s’est installé quelques jours
en juillet 2009 pour accueillir 24 de ses thésards, post-doctorants et
jeunes chercheurs ainsi que 6 chercheurs confirmés du domaine de
la « nutrition et homéostasie » pour sa 7ème école d’été.
Initié en 2003 par Xavier Leverve, cet évènement tend à sensibiliser les jeunes
scientifiques à l’analyse d’articles scientifiques afin de développer leur esprit
critique et la discussion contradictoire.
Après des sujets tels que « comportement du consommateur » en 2008 ou
encore « Evaluation du risque toxicologique des aliments » en 2007, les étu-
diants ont cette année planché sur une thématique quelque peu complexe,
mais néanmoins fondamentale pour aiguiser sa culture scientifique.
Le nombre de participants, volontairement limité, a cependant permis de
faciliter le partage de connaissances entre jeunes chercheurs et chercheurs
séniors, permettant ainsi d’aller dans le vif du sujet pour que chacun puisse
repartir « repu ». De plus l’ambiance simple et détendue vient couronner la
réussite de l’évènement que vous pourrez ressentir au fil des pages qui vont
suivre.
Si j’ai souhaité que cet opuscule soit réalisé, c’est parce qu’il m’a semblé
intéressant que la richesse des états de l’art et des discussions, puissent faire
l’objet d’une synthèse rendue accessible à tous ceux qui s’intéressent de près
ou de loin au sujet abordé.
Avant de terminer, je tiens à remercier mon adjoint Jean Fioramonti, chef
d’orchestre de cet évènement et de l’élaboration de son programme, ainsi
que Jeannine Goacolou et Sabrina Gasser pour l’important travail effectué
afin que ce document puisse voir le jour, sans oublier les intervenants pour la
qualité de leur travail, et sans qui l’école ne pourrait avoir lieu.
Vous en souhaitant une agréable lecture,
Patrick Etiévant
Chef du Département Alimentation Humaine

Comment se déroule l’école d’été ?
L’école d’été réuni doctorants, post-doctorants et jeunes chercheurs dans un lieu con-
vivial pendant 6 demi-journées, autour d’une thématique qui va leur permettre d’ap-
profondir leurs connaissances, dans un domaine plus ou moins proche de leur sujet de
thèse ou de leur activité quotidienne.
Le principe veut que, tour à tour, au cours de ces journées, les participants soient dans
la position d’enseigné et d’enseignant. Chaque thématique, déclinant le thème choisi,
s’articule autour d’une demi-journée de travail.
Tout d’abord, le chercheur senior sollicité par le département présente une synthèse de
l’état de l’art. Puis cinq articles scientifiques sélectionnés par celui-ci sont ensuite
présentés de façon critique par les doctorants sous forme de présentations orales de 10
minutes.
L’article en question, choisi et préparé au préalable par l’étudiant, donne ensuite lieu à
un débat animé par le chercheur, qui peut également aider l’étudiant dans sa présen-
tation si nécessaire.
L’objectif est donc au moins triple :
développer l’esprit critique et de la discussion contradictoire des jeunes vis-à-
vis et autour des travaux publiés dans la littérature qu’ils considèrent souvent
comme paroles d’évangile.
permettre aux jeunes de passer plusieurs journées consécutives avec des
chercheurs renommés dans les domaines traités et de leur offrir des perspec-
tives de contacts futurs.
offrir un cadre convivial, second point fort de cette école, permettant d’instau-
rer des connections fortes entre des étudiants et jeunes chercheurs qui n’au-
raient jamais eu l’occasion de se rencontrer, ainsi qu’un climat relationnel de
chercheur à chercheur avec les organisateurs et les intervenants.
Directeur de publication : Patrick Etiévant
Réalisation : Jeannine Goacolou, Sabrina Gasser
Mise en page et impression : Imprimerie Decombat

Transport et transporteurs d’acides aminés :
du métabolisme au statut redox . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Session animée par Jean-François Huneau . . . . . . . . . . . . . . . . . . . . page 5
Contrôle nerveux de l’homéostasie énergétique :
aspects centraux
Session animée par Luc Pénicaud . . . . . . . . . . . . . . . . . . . . . . . . . . page 24
Contrôle nerveux de l’homéostasie énergétique :
aspects périphériques
Session animée par Christophe Magnan . . . . . . . . . . . . . . . . . . . . . page 40
Plasticité tissulaire et cellulaire des tissus adipeux :
rôle dans l’homéostasie et l’obésité
Session animée par Louis Casteilla . . . . . . . . . . . . . . . . . . . . . . . . . . page 53
Insulino-résistance musculaire : adaptations métaboliques
et transcriptionnelles à la nutrition
Session animée par Hubert Vidal . . . . . . . . . . . . . . . . . . . . . . . . . . . page 68
Adaptations nutritionnelles et pancréas endocrine
Session animée par Bernard Portha . . . . . . . . . . . . . . . . . . . . . . . . . page 84
Sommaire
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
1
/
100
100%