Bilan d`application de la loi de bioéthique du 6 août 2004

Octobre 2008
Bilan d’application
de la loi de bioéthique
du 6 août 2004
Rapport à la Ministre de la santé,
de la jeunesse, des sports
et de la vie associative


Octobre 2008
Bilan d’application
de la loi de bioéthique
du 6 août 2004
Rapport à la Ministre de la santé,
de la jeunesse, des sports
et de la vie associative
Projet1:Mise en page 1 6/11/08 16:19 Page 1


Agence de la biomédecine - Bilan d’application de la loi de bioéthique – octobre 2008
Introduction générale
Le présent rapport sur l’application de la loi de bioéthique du 6 août 2004 s’inscrit dans le dispositif de
consultation mis en place par le Gouvernement pour aborder la révision de la loi de bioéthique du 6
août 2004.
La portée éthique des domaines couverts par la loi impliquant l’ouverture d’un débat bien préparé et
documenté, le Gouvernement a souhaité se donner les moyens d’une réflexion rigoureuse.
Le Premier ministre a d’abord saisi le Conseil d’État en vue d’une étude préalable à la révision de la
loi et a demandé au Comité consultatif national d’éthique d’identifier les problèmes philosophiques et
les interrogations éthiques, de manière à délimiter le contenu et le périmètre de la réflexion.
Pour sa part, la ministre chargée de la santé a demandé à l’Agence de la biomédecine d’établir un
bilan de l’application de la loi du 6 août 2004. Cet état des lieux vise à faire le point sur la mise en
œuvre effective de la loi au regard de l’évolution de la science et des pratiques médicales, en matière
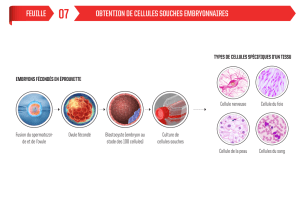
de procréation, de diagnostic ou de traitements. Il s’agit notamment de faire le point sur les recherches
sur l’embryon autorisées et sur les résultats obtenus.
Elle a également demandé à l’Agence de la biomédecine une étude de droit comparé des différentes
législations nationales pour apporter à la réflexion un éclairage international
Afin de ne pas cantonner la réflexion à un débat d’experts, la consultation de ces institutions sera
complétée par des états généraux de la bioéthique, organisés au premier semestre 2009, qui
permettront d’engager un débat public faisant appel à la participation des citoyens.
L’Agence de la biomédecine apporte donc ici sa contribution à la préparation de la révision de la loi du
6 août 2004.
Emmanuelle PRADA BORDENAVE
Directrice générale
de l’Agence de la biomédecine
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
1
/
104
100%