Génétique des syndromes myéloprolifératifs

doi: 10.1684/hma.2009.0357
Génétique des syndromes
myéloprolifératifs :
la dialectique de l’inné et de l’acquis
Genetics of myeloproliferative neoplasms:
the dialectics of innate vs acquired traits
Éric Lippert
Stéphanie Dulucq
Estelle Guérin
Laboratoire d’hématologie,
Inserm U876,
CHU de Bordeaux,
Université Victor-Segalen,
Bordeaux-II, Bordeaux
Des progrès majeurs ont été réalisés ces dernières années dans la com-
préhension de la physiopathologie des syndromes myéloprolifératifs
Philadelphie négatifs (désignés ici par SMP). La découverte d’une
mutation ponctuelle de la tyrosine kinase JAK2 (JAK2V617F) chez la
quasi-totalité des patients atteints de polyglobulie de Vaquez (PV) et
plus de la moitié des patients souffrant de thrombocytémie essentielle (TE) ou de myé-
lofibrose primitive (MP), a eu un impact important sur la prise en charge diagnostique
mais également sur la compréhension de ces maladies. Cependant, ainsi que nous
l’avons discuté dans une revue récente [1], la découverte d’une même mutation
dans trois maladies différentes a soulevé diverses questions. Certaines hypothèses,
comme celles postulant la survenue de la mutation dans un progéniteur engagé vers
la lignée érythroblastique ou mégacaryocytaire, ont été rapidement éliminées, la
mutation survenant dans une cellule-souche multipotente. D’autres, aprioricontradic-
toires, ont pourtant été renforcées, nous amenant à envisager la survenue d’un SMP et
son phénotype comme résultant de nombreux facteurs concomitants. En effet, l’hypo-
thèse selon laquelle le niveau d’expression de JAK2V617F pourrait orienter le phéno-
type est maintenant soutenue par plusieurs modèles murins de reconstitution hémato-
poïétique après infection rétrovirale des progéniteurs ou de souris transgéniques,
ainsi que par la démonstration de l’existence d’un clone homozygote pour la muta-
tion JAK2V617F chez pratiquement tous les patients atteints de PV. L’hypothèse,
apparemment alternative, de l’existence d’une mutation antérieure à la mutation de
JAK2 est maintenant confirmée, à la fois par l’étude de sous-clones présentant des
anomalies cytogénétiques différentes (acquisition de mutation JAK2V617F après
une délétion 13q ou une délétion 20q, discuté plus loin) et par la découverte de muta-
tions récurrentes dans le gène TET2 [2]. Enfin, des arguments ont aussi été accumulés
en faveur de l’hypothèse d’un déterminisme génétique.
Les SMP sont-ils génétiquement déterminés ?
La mutation V617F de JAK2 et les autres mutations découvertes ensuite dans ces mala-
dies (portant sur l’exon 12 de JAK2, sur le récepteur de la thrombopoïétine : MPL ou
plus récemment sur le gène TET2) sont des mutations acquises. Cependant, certaines
caractéristiques des SMP plaident pour un rôle du « fond » génétique. On sait depuis
plusieurs années qu’il existe des formes familiales de SMP [3, 4] : dans ces familles, les
sujets atteints peuvent présenter des SMP de phénotype identique ou parfois de phéno-
type différent (PV, TE, voire LMC ou mastocytose). Certains de ces SMP familiaux
Focus
Sous la direction de Philippe Rousselot
Hématologie 2009 ; 15 (3) : 188-93
Tirés à part :
É. Lippert
Hématologie, vol. 15, n° 3, mai-juin 2009
188
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

présentent une mutation JAK2V617F, mais dans ces cas, elle
est également acquise (restreinte aux cellules hématopoïéti-
ques). Il y a donc chez ces patients un facteur génétique trans-
mis selon un mode autosomique de pénétrance variable mais
qui ne correspond pas aux gènes habituellement remaniés
dans les SMP [3]. Récemment, l’étude de parents au premier
degré de patients porteurs de SMP dans une grande cohorte
suédoise retrouve un risque relatif supérieur à 5 de développer
un SMP [5]. Cela indique qu’au-delà des véritables formes
familialesavecuneprévalenceimportanteauseindelafamille,
les formes sporadiques ont probablement aussi des détermi-
nants génétiques. Enfin, le contexte génétique pourrait jouer
un rôle non seulement sur le risque de développer un SMP,
mais aussi sur son phénotype. Cette hypothèse a été initiale-
ment envisagée en raison des modèles murins dans lesquels
l’expression de JAK2V617F dans les cellules hématopoïétiques
entraîne constamment une polyglobulie, mais parfois une
hyperleucocytose et une fibrose plus ou moins rapide selon la
souche de souris receveuse. L’idée que le fond génétique pour-
rait modifier le phénotype a amené plusieurs groupes à étudier
certains marqueurs génétiques chez les patients porteurs de
SMP. L’une des premières études a été réalisée par le groupe
de A. Tefferi. Ces auteurs ont génotypé un certain nombre de
polymorphismes de l’ADN situés au sein ou à proximité de
gènes potentiellement importants dans la physiopathologie des
SMP [6]. Ces polymorphismes correspondent à des variations
de base dans un seul nucléotide (single nucleotide polymor-
phism : SNP, lire « snip ») et font partie des nombreuses varia-
tions de séquence identifiées dans le génome qui différencient
les individus entre eux. Chacun de ces SNP est répertorié par
un code numérique suivant les lettres rs (random snp).Onpeut
définir un allèle majeur (base la plus commune) et un allèle
mineur (base rencontrée avec une fréquence plus ou moins
grande selon la population étudiée). Chaque individu porte
deux allèles, identiques ou différents, définissant l’état constitu-
tionnel ou génotype. Les combinaisons d’allèles de plusieurs
SNP, localisés dans la même région d’ADN, définissent des
haplotypes (fragments plus ou moins longs du génome qui
ségrégent en un seul bloc lors des méioses). Ainsi, l’étude d’un
SNP chez les patients atteints de SMP, comparés à une cohorte
saine de même origine ethnique, permet de dire si un allèle est
représenté de façon conforme à ce qui est attendu dans la
population ou s’il est sur- ou sous-représenté. S’il est surrepré-
senté, il marque un plus grand risque pour les porteurs de cet
allèle de développer la maladie. Cela peut se comprendre si
la variation en question a un retentissement fonctionnel : SNP
présent dans une région codante et responsable d’un change-
ment d’acide aminé dans une séquence protéique, SNP pré-
sent dans une région promotrice et modifiant l’affinité du gène
pour tel facteur de transcription, etc. Parfois, le SNP n’a pas de
retentissement fonctionnel « en soi », mais il est le marqueur
d’un haplotype particulier dans lequel se situe un SNP fonction-
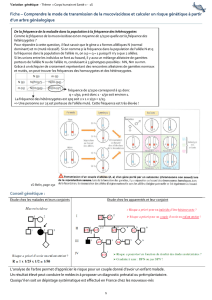
nel. Par exemple, considérons quatre SNP (figure 1).L’allèle de
référence de la position rs1 est G, l’allèle variant est A, pour le
Allèle majeur
Allèle mineur
Déséquilibre de liaison
Haplotype 1
Haplotype 2
Haplotype 3
Haplotype 4 CGTA
SNP marqueurs : rs1 et rs2/rs3/rs4
Séquence de référence
rs1 rs2 rs3 rs4
GC AT
AT CG
rs1 rs2 rs3 rs4
ATCG
ATCA
CGTG
Figure 1.Représentation schématique d’un locus comportant quatre SNP et des haplotypes possibles. rs2, 3 et 4 sont en déséquilibre de
liaison total. L’intensité de coloration de chaque losange est d’autant plus importante que le déséquilibre de liaison est plus élevé (se
reporter au texte).
Hématologie, vol. 15, n° 3, mai-juin 2009
189
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

SNPrs2:CdevientT,pourlers3:AdevientCetTdevientG
pour rs4. La séquence de référence des SNP rs1, 2, 3, 4 est
GCAT. Imaginons que dans la population de référence, le C
du rs2 soit changé pour T dans 25 % des cas. Si dans la popu-
lation étudiée (SMP), l’allèle T est retrouvé chez les trois quarts
des patients, on peut considérer qu’il est surreprésenté. Il s’agit
donc d’un allèle de prédisposition. Cependant, ce SNP ne cor-
respond pas à une région fonctionnelle du gène. Il convient
alors d’étudier des SNP à proximité afin de voir si rs2 est le
marqueur d’un haplotype. Dans la population générale, l’allèle
T de rs2 est toujours associé à l’allèle C de rs3 et à l’allèle G de
rs4. Ces allèles sont dits « en déséquilibre de liaison » et définis-
sent le même haplotype (TCG). Ce sont tous les trois des SNP
marqueurs possibles de l’haplotype. Il suffit de génotyper un
patient pour un des trois SNP pour en déduire le génotype au
niveau des deux autres SNP. Si on imagine que rs4 se trouve
en séquence codante et que son changement G pour T entraîne
une modification d’acideaminé,onpeutcomprendrealors
aisément que cette variation puisse avoir un retentissement fonc-
tionnel. Dans ce cas, on peut expliquer que la surreprésentation
de l’allèle T de rs2 soit associée à la maladie, car il est en désé-
quilibre de liaison avec l’allèle G de rs4, qui lui est susceptible
d’avoir un impact fonctionnel. Si la variation de base sur rs1
n’est pas significativement associée à celle de rs2, alors on
peut considérer que ces marqueurs ne sont pas en déséquilibre
de liaison, rs1 n’appartient donc pas à l’haplotype. Des travaux
collaboratifs récents se sont attachés à caractériser un grand
nombre de SNP à travers le génome chez des populations de
référence afin de déterminer la fréquence des différents allèles
et de définir les différents haplotypes des populations ainsi que
leur fréquence. Le projet dit « HapMap », dont les données sont
accessibles en ligne (http://www.hapmap.org/), a donc servi
de base à de nombreuses études génétiques visant à définir
des régions génétiques associées à des maladies particulières.
Ainsi, l’étude de SNP, marqueurs entourant les gènes JAK2,
MPL (récepteur de la thrombopoïétine), EPOR (récepteur de
l’érythropoïétine) et GCSFR (récepteur du G-CSF) chez des
patients atteints de PV ou de TE, permet à Pardanani et al. de
montrer que six SNP trouvés dans le locus de JAK2 sont signifi-
cativement associés au phénotype de PV plutôt que de TE.
Les SNP des autres gènes ne sont pas discriminants. Ce travail
restedescriptifetnefournitpasd’explication à cette observa-
tion, mais il renforce l’hypothèsedurôleduterraingénétique
dans le développement des SMP, hypothèse confirmée de
façon claire par trois articles parus dans le numéro d’avril de
la revue Nature genetics.
Un haplotype « 46/1 » est associé
à la survenue de la mutation
JAK2V617F
La découverte de cet haplotype associé à la survenue de la
mutation a été faite simultanément par trois équipes qui ont
mis en œuvre des stratégies différentes :
–l’équipe anglaise (laboratoire de N. Cross) a étudié (comme
Pardanani et al.) six SNP situés dans ou à proximité immé-
diate de JAK2 chez des patients dont les granuleux ont plus
de 50 % de charge allélique JAK2V617F, déterminée par
pyroséquençage [7]. Chez ces patients, l’acquisition de la
mutation JAK2V617F sur un allèle du bras court du chromo-
some 9 a été suivie d’un phénomène de recombinaison mito-
tique qui a entraîné l’échange de l’allèle sauvage de JAK2
(sur l’autre chromosome 9) avec l’allèle muté qui se retrouve
ainsi à l’état homozygote [8]. En même temps que la variation
V617F, les SNP caractéristiques de l’allèle muté se retrouvent
eux aussi « emmenés » par la recombinaison mitotique (géné-
ralement de grande taille, courant jusqu’au télomère). Si le
patient était hétérozygote pour certains de ces SNP, les
cellules ayant subi cette recombinaison se retrouvent alors
homozygotes : c’est le phénomène de perte d’hétérozygotie.
Le pyroséquençage, qui permet de détecter les variations de
séquences mais aussi de les quantifier, a été utilisé par Jones
et al. pour déterminer si un SNP était préférentiellement asso-
cié aux mutations homozygotes de JAK2. Si c’estlecas,ildoit
être représenté par un signal supérieur à 50 % chez les
patients théoriquement hétérozygotes pour ce marqueur.
De la même façon, l’allèle non muté est aussi caractérisable
s’il persiste suffisamment de cellules n’ayant pas subi la perte
d’hétérozygotie. Cette approche leur permet de constater que
chez 77 % des patients homozygotes pour la mutation
JAK2V617F, celle-ci est survenue dans le contexte d’un haplo-
type particulier dit « 46/1 ». Cet haplotype (en réalité combi-
naison de deux haplotypes : le 46 et le 1, distincts par un seul
SNP) peut être identifié par quatre SNP marqueurs, il est donc
aussi appelé « GGCC » en référence aux bases caractéristi-
ques de cet haplotype sur ces quatre SNP. Dans une popula-
tion caucasienne de référence, cet haplotype 46/1 ne repré-
sente que 24 % des haplotypes de ce bloc génomique. Il est
donc indiscutablement surreprésenté chez les patients homo-
zygotes pour JAK2V617F. De la même façon, chez les
patients porteurs de SMP hétérozygotes pour JAK2V617F
(< 50 % de charge allélique), l’haplotype 46/1 est surrepré-
senté (38 %) par rapport à une population témoin étudiée par
ce groupe (24 %) ou par le projet « HapMap ». En revanche,
cette surreprésentation n’est pas retrouvée chez les patients
porteurs d’érythrocytose idiopathique. Chez les patients hété-
rozygotes pour la mutation JAK2V617F, l’utilisation d’une
PCR spécifique de l’allèle muté, suivie de séquençage de
l’amplicon permet de déterminer si celle-ci est survenue sur
un allèle porteur de l’haplotype 46/1. C’est le cas pour
74 % des patients, tandis que cet haplotype n’est retrouvé
associé à l’allèle sauvage de JAK2 que chez 12 % des
patients. Ces éléments démontrent clairement une association
entre l’haplotype 46/1 et la survenue d’une mutation de
JAK2. De façon intéressante, les SNP trouvés surreprésentés
dans les PV par rapport aux TE par l’équipe de Tefferi sont
aussi des marqueurs de l’haplotype 46/1, non caractérisé
comme tel alors ;
Hématologie, vol. 15, n° 3, mai-juin 2009
190
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

–l’équipe de R. Levine arrive à des résultats semblables [9]
en ayant mis en œuvre une stratégie identique à celle de
Paradanani et al. (comparaison PV vs TE), ne reposant plus
sur l’étude de quelques SNP sélectionnés selon une appro-
che de « gène candidat », mais en étudiant la globalité du
génome à l’aide de puces (SNP array). Ces outils permettent
l’étude de près de 250 000 SNP répartis sur tout le génome.
Cette large couverture du génome a permis à ces auteurs de
mettre en évidence quatre régions potentiellement intéressan-
tes, car présentant des fréquences génotypiques significative-
ment différentes entre PV et TE, situées en 4q31, 7p11,
9p24 et 3q21. Le SNP étudié en 9p24 (rs10974944) cor-
respond à un des quatre SNP marqueurs de l’haplotype
46/1 ou GGCC. L’allèle G trouvé surreprésenté par Kilpi-
vaara et al., à la fois dans le groupe PV vs TE mais aussi
dans le groupe des SMP vs population générale, définit en
effet ce même haplotype 46/1 et constitue de par cette sur-
représentation un facteur de risque de développer un SMP
JAK2 muté ;
–enfin, c’est par une approche tout à fait distincte que
Olcaydu et al. ont découvert, eux aussi, l’importance de
cette prédisposition génétique. Ces auteurs avaient postulé
l’existence de mutations antérieures à JAK2V617F en raison
de l’existence chez certains patients atteints de SMP, de clo-
nes porteurs d’anomalies génétiques acquises (délétion du
bras long du chromosome 20) dont la représentativité était
plus importante que celle des cellules mutées sur JAK2 [10].
Cette observation suggérait donc fortement que la mutation
JAK2V617F était survenue dans une cellule déjà porteuse de
délétion 20q. Afin d’affiner cette hypothèse, les auteurs ont
tenté de reconstituer l’histoire de l’acquisition des mutations
chez des patients présentant des anomalies génétiques multi-
ples caractérisables en biologie moléculaire (délétion 20q,
délétion 13q, perte d’hétérozygotie 9p). Pour ce faire, des
colonies issues d’un progéniteur unique ont été caractérisées,
amenant à la conclusion que chez certains patients, la muta-
tion JAK2V617F a été acquise deux fois de façon indépen-
dante. Par exemple, chez un même patient, la mutation a pu
être acquise une fois dans un clone présentant une del(20q) et
une autre fois dans un clone présentant une del(13q)
(figure 2A). Cette découverte importante suggère qu’il existe
une « hypermutabilité » chez les patients atteints de SMP, que
del(13q)
del(13q)
+JAK2
V617F
del(20q)
del(20q)
+JAK2
V617F
JAK2-1849
rs12343867
TC
T
G
JAK2-1849
TT
C
G
AB
rs12343867
JAK2-1849
GT
C
G
rs12343867
Figure 2.Acquisitions multiples de la mutation JAK2V617F. A) Au sein de l’hématopoïèse clonale (clone représenté par un triangle noir)
survient une délétion 13q (sous-clone rouge) et une délétion 20q (sous-clone gris). Une mutation JAK2V617F peut ensuite avoir lieu
dans chacun des sous-clones, donc deux fois, de façon indépendante ; B) Au sein d’une hématopoïèse clonale chez un sujet hétérozy-
gote pour le SNP12343867, la mutation JAK2G1849T (V617F) peut se produire sur l’allèle C du SNP (sous-clone représenté par un trian-
gle rouge) ou sur l’allèle T (sous-clone représenté par un triangle gris). L’association est détectable par une PCR spécifique d’allèle.
Hématologie, vol. 15, n° 3, mai-juin 2009
191
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

cette hypermutabilité est antérieure à la survenue de la muta-
tion JAK2V617F et que JAK2 est une cible particulièrement
fréquente de ces mutations. Afin d’étendre cette étude aux
patients qui n’ont pas d’anomalie cytogénétique caractéris-
tique, les auteurs ont développé une PCR spécifique d’allèle
JAK2V617F couplée à la caractérisation d’un SNP situé à
proximité [11]. Le génotypage du SNP est assuré par l’utilisa-
tion de sondes d’hydrolyse fluorescentes (de type Taqman)
spécifiques de l’allèle C ou de l’allèle T du SNP
rs12343867. Ainsi, chez les patients hétérozygotes pour ce
SNP, on s’attend à ce que la PCR spécifique de l’allèle sau-
vage de JAK2 soit toujours associée à la détection du SNP
caractéristique de l’allèle sur lequel la mutation est survenue,
disons, l’allèle T pour rs12343867, tandis que la PCR spéci-
fique de l’allèle muté de JAK2 sera associée à l’allèle C de
rs12343867 (figure 2B). Chez 2,8 % des patients informatifs,
cette discrimination n’est pas totale, ce qui indique que la
mutation JAK2V617F est survenue deux fois, une fois sur un
allèle porteur d’un C en rs12343867 et une fois sur un allèle
porteur d’un T. Considérant que la mutation peut aussi surve-
nir deux fois sur le même allèle, on peut en déduire que cette
mutation est acquise dans plusieurs clones au cours de l’his-
toire de plus de 2,8 % des SMP. Au cours de ces recherches,
ces auteurs notent aussi que chez 85 % des patients, la muta-
tion survient sur l’allèle C de rs12343867, alors que cet allèle
n’est attendu que dans 25 % des cas. La comparaison de 120
patients porteurs de SMP JAK2 sauvage à 213 patients
JAK2V617F+ confirme que l’allèle C entraîne une prédisposi-
tion à la mutation JAK2. L’élargissement de l’étude aux SNP
voisins permet de conclure, comme dans l’étude de Jones et
al., à la surreprésentation de l’haplotype 46/1 chez les
patients présentant une mutation JAK2V617F, en particulier
àl’état homozygote.
Pourquoi l’haplotype 46/1
confère-t-il une susceptibilité
aux SMP ?
Comme indiqué dans l’article de Jones et al., deux mécanis-
mes peuvent expliquer cette association :
–la mutation JAK2V617F survient indifféremment sur tous les
haplotypes, mais 46/1 est en déséquilibre de liaison avec
un facteur interagissant avec JAK2, favorisant donc la surve-
nue d’une maladie ayant un retentissement clinique ;
–46/1 présente une particularité qui favorise la mutation du
gène JAK2. Les données « HapMap » indiquent que JAK2
est situé dans un bloc haplotypique d’environ 300 kb com-
portant aussi les gènes INSL4 et INSL6, mais ceux-ci ne sont
pas exprimés dans les cellules hématopoïétiques. Jones et al.
penchent pourtant pour l’hypothèse d’un facteur fonctionnel
favorisant le développement de la maladie. Ils étudient donc
la pousse de colonies granulomonocytaires et érythroïdes à
partir du sang de 56 individus sains et observent que les
porteurs de l’haplotype 46/1 ont significativement moins de
CFU-GM mais un nombre identique de BFU-E. Il pourrait
donc y avoir une différence dans la « signalisation JAK2 »
chez ces sujets. Cela pourrait aussi expliquer que cet haplo-
type est significativement associé au développement de la
maladie de Crohn, maladie dans laquelle la fonction lym-
phocytaire, partiellement dépendante de JAK2, est impor-
tante. Cependant, la production d’ARN de JAK2 produit à
partir de l’allèle 46/1 est identique à celle de l’allèle sau-
vage lorsque les deux sont quantifiées par pyroséquençage
d’un SNP de la région codante. Par ailleurs, Olcaydu et al.
soulignent que deux SNP présents dans la région promotrice
de JAK2 ne sont pas en déséquilibre de liaison, éliminant
l’hypothèse d’une modification d’expression par des poly-
morphismes du promoteur. De plus, chez deux individus
ayant acquis la mutation V617F sur deux allèles : une dans
le contexte de l’haplotype 46/1 (GGCC) et l’autre dans un
autre contexte (TCTT), le clone TCTT prédomine sur le clone
GGCC dans les deux cas, ce qui va à l’encontre d’un avan-
tage pour les cellules mutées sur 46/1. Ces auteurs propo-
sent donc que la séquence de l’haplotype 46/1 soit respon-
sable d’une « hypermutabilité locale ». Le mécanisme sous-
jacent n’est pas connu, mais il est intéressant de noter que
des cas semblables ont été décrits avec un allèle de suscep-
tibilité aux mutations d’APC retrouvé avec une grande fré-
quence dans les cancers colorectaux dans une population
juive ashkénaze ou de mutation de p53 dans les cancers
bronchiques dans une population d’Américains d’origine
africaine.
Au-delà de l’hétérogénéité phénotypique des SMP, on voit
donc que les mécanismes physiopathologiques de ces mala-
dies sont eux aussi divers et complexes. Contrairement au cas
« simple » de la leucémie myéloïde chronique où une maladie
correspond à un gène de fusion, les intervenants semblent
plus nombreux dans les SMP. Si la mutation JAK2V617F est
un élément majeur de la physiopathologie de beaucoup
d’entre eux, d’autres mutations acquises et des variations
génétiques jouent un rôle indiscutable qui fera, n’en doutons
pas, l’objet de passionnantes études à venir. ■
RÉFÉRENCES
1.Gue
´rin E, Praloran V, Lippert E. Les mutations de JAK2 dans les
syndromes mye
´loprolife
´ratifs en 2008. He
´matologie 2008 ; 14
368-7.
2.Delhommeau F, Dupont S, James C, et al. TET2 is a novel tumor
suppressor gene inactivated in myeloproliferative neoplasms: iden-
tification of a pre-JAK2 V617F event [Abstract]. Blood 2008 ;
112 : 1ba-3ba.
3.Kralovics R, Buser AS, Teo SS, et al. Comparison of molecular
markers in a cohort of patients with chronic myeloproliferative
disorders. Blood 2003 ; 102 : 1869-71.
4.Bellanne
´-Chantelot C, Chaumarel I, Labopin M, et al. Genetic
and clinical implications of the Val617Phe JAK2 mutation in 72
families with myeloproliferative disorders. Blood 2006 ; 108 :
346-52.
Hématologie, vol. 15, n° 3, mai-juin 2009
192
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.
 6
6
1
/
6
100%