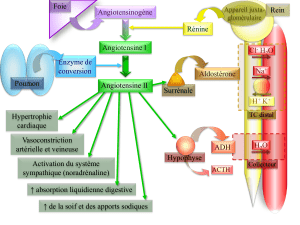
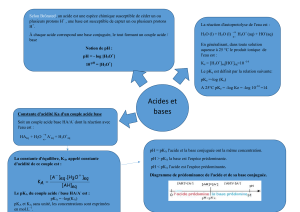
Chapitre 3. Transferts de protons

© Jacques Penelle
21
Chapitre 3. Transferts de protons
3.1. Introduction
En chimie, on qualifie de "proton" indifféremment l'espèce H+ et l'espèce H3O+. La
première est, comme présenté dans la section d’atomistique, une particule élémentaire
constituant du noyau. La notation classique des physiciens pour cette particule est p+.
Le noyau de l'atome d'hydrogène n'étant constitué que d'un proton, on a adopté en
chimie la notation H+. L'espèce H+ peut être observée en phase gazeuse, mais sa
grande affinité pour les électrons la fait réagir rapidement avec les atomes porteurs de
paire libre pour former un lien covalent. Dans l'eau, le seul solvant considéré lors de cet
exposé, on a donc :
H
O H
H
O H
H
(+)
H+
ou, en notation plus classique :
H+ + H2O -> H3O+
H3O+ est donc la seule espèce "proton" observable en solution aqueuse. Lorsque l'on
parle de proton en solution aqueuse, on s’y réfère donc automatiquement. Par contre,
lorsque l'on parle de transfert de proton, on parle bien de l'espèce H+ qui est transférée
d'une espèce moléculaire vers une autre espèce moléculaire, comme par exemple :
H3O+ + NH3 -> H2O + NH4+
On appellera acide toute espèce moléculaire capable de céder un proton et base toute
espèce moléculaire capable d'accepter un proton (définition dite de "Brönsted").
3.2. Force des acides et des bases
3.2.1. Réactions de transfert impliquées
L'eau est à la fois une base et un acide. Cette double propriété entraîne quelques
conséquences fondamentales en ce qui concerne les transferts de protons dans l'eau :
(a) L'eau réagit avec elle-même selon l'équation :
H2O + H2O OH- + H3O+

© Jacques Penelle
22
Cette réaction est appelée réaction d'autoprotolyse (on trouvera parfois aussi le
terme d’auto-ionisation dans la littérature scientifique). Elle est équilibrée et
fortement déplacée vers les réactifs. La constante d'équilibre correspondante
(constante d'autoprotolyse) est généralement notée Kw (le w trouvant son origine
dans l'anglais water et dans l'allemand Wasser). On la définit par la relation :
Kw = [OH-].[H3O+] (3-1)
la contribution des réactifs ([H2O]2) étant omise au dénominateur dans la
mesure où l'eau est aussi le solvant de la réaction et sa concentration de 55,5 M
reste constante (le taux de conversion est en effet d’approximativement
0,00000018%).
A 25°C et sous 1 atmosphère, la valeur numérique de Kw est de 10-14!; à 40°C,
température plus proche de celles de beaucoup de mammifères, elle vaut
10-13,534. Sa valeur exacte sous diverses conditions expérimentales peut être
trouvée dans des ouvrages spécialisés. Dans le cadre de ce cours, nous
utiliserons la valeur 10–14.
(b) Tout acide (donneur de proton noté A-H) sera caractérisé par sa capacité à
réagir avec l'eau selon l'équation :
A-H + H2O A- + H3O+
(remarque : il existe des acides chargés positivement (NH4+ par exemple) ;
dans ce cas, on écrira l'équation générale sous la forme!:
A-H+ + H2O A + H3O+)
L'espèce moléculaire obtenue après perte du proton (A- ou A) est appelée base
conjuguée.
(c) Par analogie, toute base (accepteur de proton) sera caractérisée par sa capacité
à réagir avec l'eau selon l'équation :
A- + H2O A-H + OH-
ou
A + H2O A-H+ + OH-
L'espèce moléculaire obtenue après acceptation du proton (A-H ou A-H+) est
appelée acide conjugué.

© Jacques Penelle
23
3.2.2. Classement qualitatif des acides et des bases
Les acides et les bases peuvent être divisés en deux catégories :
(1) Les acides forts et les bases fortes sont caractérisés par leur capacité à réagir
de manière complète avec l'eau. Pour ces espèces, on a donc respectivement les
équations :
A-H + H2O -> A- + H3O+
ou
A- + H2O -> A-H + OH-
Les acides forts les plus courants sont HI, HBr, HCl, HClO4, HClO3, HNO3 et
H2SO4 (ce dernier, uniquement pour le don de son premier proton). Les seuls
acides forts de la biochimie sont les dérivés de l’acide sulphonique (-SO3H, voir
section «!chimie organique!») comme la taurine.
Les bases conjuguées des acides forts sont donc incapables de réagir avec l'eau
par transfert de proton. Lorsque l’on dissout du chlorure de sodium
(constituant majeur du sel de cuisine) dans l'eau, on ne forme donc pas d'espèce
OH- par la réaction :
Cl- + H2O HCl + OH-
(2) Les acides et les bases faibles, au contraire, atteignent un équilibre lorsqu'ils
réagissent avec l'eau. Une fraction plus ou moins importante de l'acide ou
de la base de départ restera présente en solution et en équilibre avec l'espèce
conjuguée.
3.2.3. Classement quantitatif des acides et des bases faibles
Il est possible de déterminer une constante d'équilibre pour la réaction des acides et des
bases faibles avec l'eau.
La constante d'acidité (Ka) est définie par la réaction :
HA + H2O H3O+ + A-
On a donc pour la constante d’équilibre!:
!
Ka=
H3O+
[ ]
A"
[ ]
HA
[ ]
(3-2)
Remarque : [H2O] est à nouveau omis pour la même raison que celle mentionée
précédemment pour Kw.

© Jacques Penelle
24
On utilise souvent une échelle logarithmique, l'échelle p :
Par définition, p = - log d'où pK = - log K
On a aussi : pH = - log [H3O+] (3-3)
Par exemple,
pKa = 4,5 ! Ka = 10-4,5
Il faut toujours bien se rappeler que l'échelle p est une échelle logarithmique en base 10.
On a donc par exemple :
pH
[H3O+]
1
10-1 M = 0,1 M
2
10-2 M = 0,01 M
En passant d'un pH de 1 à 2, on diminue donc la concentration en protons d'un facteur
10.
La constante de basicité (Kb) est définie par :
A- + H2O HA + OH-
d’où
!
Kb=
HA
[ ]
OH"
[ ]
A"
[ ]
(3-4)
On peut passer de Kb à Ka ou l'inverse par la relation :
Ka . Kb =Kw (3-5)
Démonstration :
!
Ka.Kb=
H3O+
[ ]
A"
[ ]
HA
[ ]
HA
[ ]
OH"
[ ]
A"
[ ]
Ka.Kb=H3O+
[ ]
OH"
[ ]
Ka.Kb=Kw
La détermination expérimentale des pKa permet l'établissement d'une échelle d'acidité
dans l'eau. De nombreuses valeurs peuvent être trouvées dans des compilations
spécialisées. Une échelle résumée au couples acido-basiques les plus courants est
disponible au tableau 3-1 ci-dessous. Elle reprend les valeurs de pKa dans l'eau à 25°C
pour divers couples "acide + base conjuguée". Deux couples essentiels correspondant
aux couples de H2O en tant qu'acide et que base (lignes 7 et 28) définissent les
"frontières" de la zone couverte par les couples acides-bases faibles.

© Jacques Penelle
25
Tableau 3-1. Valeurs de pKa pour quelques couples acido-basiques caractéristiques
(à 298 K)
forme acide
pKa
base conjuguée
1
acide chlorhydrique
HCl
Cl-
2
acide bromhydrique
HBr
Br-
3
acide iodhydrique
HI
I-
4
acide sulfurique (1)
H2SO4
HSO4
-
5
acide perchlorique
HClO4
ClO4
-
6
acide nitrique
HNO3
NO3
-
limite des acides forts
7
ion hydronium
H3O+
H2O
8
acide oxalique (1)
H2C2O4
1,2
HC2O4
-
9
acide sulfurique (2)
HSO4-
2,0
SO4
2-
10
acide phosphorique (1)
H3PO4
2,2
H2PO4
-
11
glycine (1)
C2H6NO2
+
2,3
C2H5NO2
12
acide pyruvique
C3H4O3
2,4
C3H3O3
-
13
acide fluorhydrique
HF
3,2
F-
14
acide formique
CH2O2
3,8
CHO2
-
15
acide oxalique (2)
HC2O4
-
4,2
C2O4
-
16
acide acétique
C2H4O2
4,8
C2H3O2
-
17
ion pyridinium
C5H6N+
5,3
C5H5N
18
acide carbonique (1)
H2CO3
6,4
HCO3
-
19
acide sulfhydrique (1)
H2S
7,1
HS-
20
acide phosphorique (2)
H2PO4
-
7,2
HPO4
2-
21
acide cyanhydrique
HCN
9,2
CN-
22
ion ammonium
NH4
+
9,3
NH3
23
acide borique (1)
H3BO3
9,3
H2BO3
-
24
glycine (2)
C2H5NO2
9,8
C2H4NO2
-
25
acide carbonique (2)
HCO3
-
10,3
CO3
2-
26
ion methylammonium
CH6N+
10,6
CH5N
27
acide phosphorique (3)
HPO4
2-
12,3
PO4
3-
28
eau
H2O
OH-
limite des bases fortes
29
ammoniac (-> amidure)
NH3
NH2
-
30
acide sulfhydrique (2)
HS-
S2-
acide
conjugué
forme basique
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
1
/
24
100%