CINÉTIQUE DES RÉACTIONS REDOX

Cinétique des réactions redox page 1/6
CINÉTIQUE DES RÉACTIONS REDOX
I-1) Pour le couple Cu
2+
/Cu
(S)
dans l’eau pure, la demi équation redox s’écrit Cu
2+
+ 2e
–
= Cu
(S)
donc le potentiel est
( )
2+
2+
(S)
0,059 [Cu ]
Cu / Cu log
2
E E c
= ° +
°
.
En présence d’ammoniac, il se forme le complexe Cu(NH
3
)
42+
suivant le bilan
Cu
2+
+ 4NH
3
= Cu(NH
3
)
42+
.
Si β est la constante de formation de ce complexe, la condition d’équilibre s’écrit
2 4
3 4
2+ 4
3
[Cu(NH ) ]
[Cu ][NH ]
c
+
°
β = et le potentiel peut s’écrire
( )
[ ]
2+ 3
2+ 3 4
(S) 4
3
[Cu(NH ) ]
0,059
Cu / Cu log
2NH
c
E E
°
= ° +
β
.
Mais pour le couple Cu(NH
3
)
42+
/Cu
(S)
, la demi équation redox s’écrit
Cu(NH
3
)
2+
+ 2e
–
= Cu
(S)
+ 4NH
3
et le potentiel s’écrit
( )
[ ]
2+ 3
2+ 3 4
3 (S) 4
3
[Cu(NH ) ]
0,059
Cu(NH ) / Cu log
2NH
c
E E
°
= ° +
.
En identifiant les deux expressions du potentiel correspond à des couples de mêmes états
redox (+II/0), il vient E°(Cu(NH
3
)
42+
/Cu
(S)
) = E°(Cu
2+
/Cu
(S)
) – 0,03log(β).
Comme β > 1 puisque le complexe Cu(NH
3
)
2+
est stable, on a
E°(Cu(NH
3
)
42+
/Cu
(S)
) < E°(Cu
2+
/Cu
(S)
) .
Conclusion : Le cuivre est donc plus réducteur en présence d’ammoniac que dans l’eau
pure.
2) Si l’électrode de cuivre fonctionne en anode, le cuivre est oxydé en Cu
2+
. Il n’y a pas de
courant limite de diffusion puis le réducteur (Cu) est toujours présent dans l’anode. L’autre
oxydation possible (H
2
O en O
2
) ne se fait pas car le potentiel correspondant est plus élevé.
Si l’électrode de cuivre fonctionne en cathode dans
l’eau pure, il se produit la réduction O
2(G)
→ H
2
O et non
H
2
O → H
2(G)
dont le potentiel est plus faible que celui du
couple du cuivre. Il existe un courant limite de diffusion dû à la
diffusion des molécules de O
2
vers l’électrode de cuivre.
La position relative des deux courbes montre qu’il
n’existe pas de valeur du potentiel pour lequel il existe un point
de fonctionnement sur les deux courbes tel que i
C
= – i
A
. La
vitesse de la réaction d’oxydation du cuivre par O
2(G)
est donc
nulle.
Conclusion : le cuivre n’est pas oxydé par l’oxygène dans une eau aérée à cause d’un
empêchement cinétique, bien que E°(Cu
2+
/Cu
(S)
) < E°(O
2
/H
2
O).
3) Le cuivre est plus réducteur en présence de NH
3
d’après la première question. On peut
donc prévoir que la courbe anodique relative au processus Cu
(S)
→ Cu(NH
3
)
42+
est déplacée vers
la gauche. On a donc l’allure suivante :
Il existe un point de fonctionnement sur les deux
courbes tel que i
C
= – i
A
: le cuivre est oxydé par O
2
suivant le
bilan Cu
(S)
+
1
2
O
2(G)
+ 4NH
3
+ H
2
O = Cu(NH
3
)
42+
+ 2OH
–
.
i (en A)
E
(en V)
Cu
(S)
Cu
2+
O
2
(sur cuivre)
H
2
O
i (en A)
E
(en V)
Cu
(S)
Cu(NH
3
)
4
2+
O
2
(sur cuivre)
H
2
O
i
A
i
C

Cinétique des réactions redox page 2/6
La solution obtenue, de couleur bleue foncée à cause de la présence du complexe
Cu(NH
3
)
42+
, est appelée liqueur de Schweitzer. Elle possède des propriétés oxydantes et dissout la
cellulose qui est insoluble dans les solvants usuels.
________________________________________________________________________________
II-1) Les deux systèmes étant rapides, on peut prévoir l’allure des courbes de la figure a.
2) Le potentiel du couple PbO
2
/PbSO
4
est supérieur à celui de Pb SO
4
/Pb donc l’oxydant
PbO
2
oxyde spontanément le réducteur Pb en mode pile. La courbe anodique concerne donc le
couple PbSO
4
/Pb et la courbe cathodique le couple PbO
2
/Pb SO
4
. Les courbes I = f(V) dans ce mode
sont donc celles de la figure b.
Comme les systèmes sont rapides, il n’y a pas de surtension. La fem de la pile qui ne débite
pas est donc U = E
CATH
– E
ANOD
soit numériquement U = 1,69 – (–0,13) = 1,82 V. Quand la pile
débite, la tension est un peu plus faible.
3) En mode électrolyseur, on force l’oxydation de Pb
2+
en PbO
2
à l’anode et sa réduction en
Pb à la cathode. Les courbes I = f(V) dans ce mode sont donc celles de la figure c. Pour définir plus
précisément le point de fonctionnement, il faudrait connaître l’intensité.
4) L’eau qui sert de solvant à l’acide sulfurique.
On peut envisager sa réduction (en H
2(G)
) et son
oxydation de en O
2(G)
. Si ces réactions ne se produisent
pas, c’est à cause de surtensions. On peut prévoir l’allure
des courbes ci-contre.
________________________________________________________________________________
III-Par temps humide, une pellicule d’eau se forme au niveau de la rayure. Cette
couchecontient du dioxygène dissous qui peut jouer le rôle d’oxydant et elle
permet la diffusion des ions formés lors d’un processus électrochimique.
Puisque l’on observe la corrosion du fer, c’est qu’il y a formation de Fe
2+
:
le fer constitue l’anode où se produit l’oxydation.
La réduction de O
2
se produit nécessairement sur le chrome,
qui joue le rôle de cathode, selon le processus
1
2
O
2(G)
+ H
2
O + 2 e
–
⇄ 2 OH
–
.
Puisque la réaction rédox a bien lieu, c’est que la disposition
des courbes intensités potentiel est la suivante : il existe un potentiel
mixte et une intensité de corrosion.
L’ensemble fer, chrome, eau aérée constitue
donc une pile que l’on peut schématisée ainsi :
Remarque : la réduction de O
2
entraîne la
formation de OH
–
: le milieu devient basique.
Par ailleurs, les ions Fe
2+
sont oxydés en Fe
3+
dont l’hydroxyde Fe(OH)
3(S)
précipite facilement. Il est
de couleur jaune-rouge. Lorsqu’il se déhydrate,
l’oxyde de Fe(III) correspondant forme la rouille.
Pb
→
PbSO
4
PbSO
4
←
PbO
2
V
I
–0,13
1,69
figure b
PbSO
4
→
PbO
2
Pb
→
PbSO
4
PbSO
4
←
PbO
2
V
I
–0,13
1,69
Pb
←
PbSO
4
figure a
PbSO
4
→
PbO
2
V
I
–0,13
1,69
Pb
←
PbSO
4
figure c
I
A
I
C
=
–
I
A
PbSO
4
→
PbO
2
V
I
–0,13
1,69
Pb
←
PbSO
4
I
A
I
C
=
–
I
A
H
2
O
→
O
2(G)
H
2(G)
←
H
2
O
i (en A)
E
(en V)
Fe
(S)
Fe
2+
O
2
(sur
chrome)
H
2
O
i
COR
–
i
COR
Fe
(S)
Cr
(S)
⊕
–
I
Fe
2+
O
2(G)
H
2
O
Cr
Cr
Fe

Cinétique des réactions redox page 3/6
Conclusion : la corrosion du fer est favorisée par la présence du chrome au niveau des
rayures. Celui-ci n’apporte donc qu’une protection de type « isolation spatiale ».
________________________________________________________________________________
IV-1-a) corrosion au niveau de la ligne de flottaison :
Plus on descend dans la l’eau, plus la concentration de O
2
dissous
diminue. La surface de fer de la coque se comporte alors comme un
ensemble de micropiles d’Evans crées par les différences de concentration
de O
2
.
dans les zones plus riches en O
2
, celui-ci se réduit suivant le processus
électrochimique :
1
2
O
2(G)
+ 2H
2
O + 2e
–
→ 2 OH
–
. Cette zone de fer joue le rôle de cathode.
dans les zones plus pauvres en O
2
, il y a oxydation du fer suivant le processus
Fe
(S)
→ Fe
2+
+ 2e
–
. Cette zone de fer joue le rôle d’anode.
C’est le cas au voisinage de la ligne de flottaison car elle correspond à l’interface
eau/atmosphère et il y a donc de l’oxygène dissous dans l’eau.
corrosion au niveau de l’hélice :
Le cuivre est moins réducteur que le fer d’après le potentiel du couple Cu
2+
/Cu
(S)
donc le fer
joue le rôle d’anode et s’oxyde en Fe
2+
pendant que le cuivre est la cathode où O
2
se réduit en OH
–
.
Le passage des électrons entre anode et cathode est assuré par le contact directe entre les métaux. Il
faut donc l’éviter.
On peut prévoir la disposition des courbes intensités potentiel suivante, dans les deux cas
étudiés ci-dessus :
b) L’eau de mer contient des ions Na
+
et Cl
–
qui sont porteurs de charge. Elle est
donc plus conductrice que l’eau douce ce qui favorise les processus électrochimiques par fermeture
des courants électriques.
2) l’inhomogénéité de la concentration de O
2
ne peut être évitée ou modifiée. Pour empêcher la
corrosion, il faut isoler le fer de l’eau de mer par des
couches de peinture sans rayure.
on peut empêcher la corrosion de la coque en
plaçant dessus un métal plus réducteur que le fer. On utilise pour cela une anode sacrificielle
généralement en zinc comme cet exemple commercial. Elle est placée contre la coque.
________________________________________________________________________________
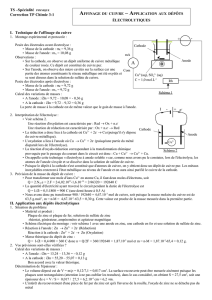
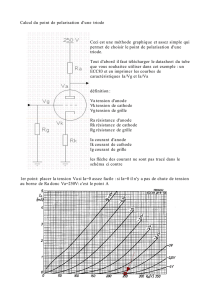
V-1-a) À l’anode se produisent les oxydations. Compte tenu des espèces présentes, on peut
envisager les processus suivants :
3H
2
O → 2H
3
O
+
+
1
2
O
2(G)
+ 2e
–
avec E
1
= 1,23 – 0,06pH soit ici E
1
= 0,93 V à
pH = 5 ;
NH
4+
peut être oxydé en NO ou NO
2
dans certaines conditions mais cela n’est pas
pris en compte par l’énoncé.
Poids de zinc (en Kg) 6,7
Poids de l'anode (en Kg) 7
Longueur (en mm) 233
Largeur (en mm) 132
Épaisseur (en mm) 57
i (en A)
E
(en V)
Fe
(S)
Fe
2+
O
2
(sur fer)
H
2
O
i
COR
–
i
COR
près de la ligne de flottaison
i (en A)
E
(en V)
Fe
(S)
Fe
2+
O
2
(sur cuivre)
H
2
O
i
COR
–
i
COR
près de l’hélice
Fe
O
2

Cinétique des réactions redox page 4/6
2SO
42–
→ 2S
2
O
82–
+ 2e
–
E
2
= E° = 2,08 V.
À la cathode se produisent les réductions. Compte tenu des espèces présentes, on peut
envisager les processus suivants :
2H
2
O + 2e
–
→ H
2(G)
+ 2OH
–
E
3
= 0 – 0,06pH soit ici E
3
= –0,30 V à pH = 5 ;
Mn
2+
+ 2e
–
→ Mn
(S)
E
4
= –1,17 V.
b) Du seul point de vue de la thermodynamique (c’est-à-dire en ne considérant que
les valeurs des potentiels des couples), la réaction la plus facile met en jeu le réducteur du couple
anodique de potentiel le plus bas (ici H
2
O) avec l’oxydant du couple cathodique de potentiel le plus
haut (ici H
2
O) suivant le bilan H
2
O = H
2(G)
+
1
2
O
2(G)
.
La tension minimale à appliquer est donc E = E
A
– E
C
= E
1
– E
3
. A.N. E = (0,93) – (–0,30)
= 1,23 V.
2) Si l’on obtient du manganèse à la cathode, c’est que la surtension de réduction de H
2
O sur
la cathode est élevée de façon que E
3
’ < E
4
. La tension minimale à appliquer est donc dans ce cas
E = E
A
– E
C
= E
1
– E
4
soit numériquement E = (0,93) – (–1,17) = 2,10 V.
3) La chute ohmique de potentiel s’ajoute à la différence de potentiel chimique donc
U
+
– U
–
= (E
A
+ η
A
) – (E
C
+ η
C
) +RI.
A.N. U
+
– U
–
= [(0,93) + (0,90)] – [(–1,17) + (–0,20) + 1,25 = 4,45 V.
4-a) D’après ce qui précède, le bilan de l’électrolyse est
Mn
2+
+ 3H
2
O = Mn
(S)
+ 2H
3
O
+
+
1
2
O
2(G)
.
Pendant une journée de durée τ où circule un courant constant I, il passe la charge q = Iτ,
soit une quantité de matière d’électrons
A
q
n
N e
=
I
τ
=
F
. D’après le processus de réduction des Mn
2+
écrit ci-dessus, la quantité de manganèse formée est
Mn
2 2
n I
n
τ
= =
F
si le rendement faradique est
unitaire. La masse de manganèse obtenue est donc
Mn Mn
2
I
m M
τ
=
F
.
A.N.
3
3
Mn
(35 10 )(86400)
(55 10 )
2(96500)
m
−
×
= ×
= 862 kg.
b) À la cathode se produit la réduction
de H
2
O (ou H
3
O
+
) en plus de celle de Mn
2+
. Une
partie des électrons qui circulent ne servent donc pas
à produire du manganèse. On a le schéma ci-contre :
Par définition, le rendement faradique est
REELLE
F
Mn
(Mn)
m
m
ρ =
soit numériquement
F
530
862
ρ =
= 0,615. Seulement 61,5% de l’intensité circulant
dans l’électrolyseur sert à produire le manganèse.
c) Le courant étant constant, la
puissance reçue par l’électrolyseur est
P
= (
U
+
–
U
–
).
I
et l’énergie consommé pendant la durée
τ
est
W
=
P
.τ
. En tenant compte du rendement
faradique, la masse de manganèse produite pendant cette durée est
Mn F Mn
2
I
m M
τ
= ρ
F
. En reportant,
i
0,93
η
Α
U (en V)
H
2
OO
2
Mn
(S)
Mn
2+
I
C
I
A
η
C
i
C
total
H
3
O
+
H
2(G)

Cinétique des réactions redox page 5/6
on obtient
Mn
F Mn
2
( )
m
W U U M
+ −
= − ρ
F
soit, par unité de masse de manganèse produit,
F Mn
2
( )
w U U M
+ −
= − ρ
F
A.N.
3
2(96500)
(4,45)
(0,615)(55 10 )
w−
=×
= 25,4
×
10
6
J.kg
–1
soit
6
25,4 10
3600
×
= 7,05 kWh.kg
–1
.
________________________________________________________________________________
VI-1) À l’anode se produit l’oxydation suivant le
processus Cu
(S)
→
Cu
2+
+ 2
e
–
.
À la cathode se produit la réduction suivant le
processus Cu
2+
+ 2
e
–
→
Cu
(S)
.
Le point de fonctionnement de l’électrolyseur est
fixé par la valeur de
∆U
telle que
I
C
= –
I
A
. On a donc le
schéma ci-contre.
Si le potentiel cathodique devient trop faible, il
se produit la réduction de H
2
O en O
2
sur l’électrode de cuivre, ce qui diminue le rendement de
l’électrolyse puisque des électrons ne servent plus à transformer Cu
2+
en Cu
(S)
. La tension doit donc
restée inférieure à 0,34 – 0 soit environ 0,3 V.
Remarque : la tension réellement imposée aux bornes de la cuve est
U
+
–
U
–
=
∆U
+
RI
A
où
R
est la résistance de l’électrolyseur.
2) D’après les valeurs des potentiels standard des couples que l’on peut supposer rapides
puisqu’ils mettent en jeu un métal comme réducteur, on
peut prévoir l’allure des courbes intensité-potentiel
suivante :
À l’anode, les traces de Fe
(S)
sont oxydées plus
facilement que Cu
(S)
mais pas celles de Ag
(S)
car la
courbe d’oxydation de Fe est « à gauche » de celle de
Cu alors que celle de Ag est « à droite ».
À la cathode, Cu
2+
est plus facilement réduit
que Fe
2+
car sa courbe de réduction est « à droite » de
celle de Fe
2+
.
L’électrolyse élimine donc le fer de l’anode par oxydation sans qu’il se redépose ensuite sur
la cathode par réduction. Les atomes d’argent libérés par destruction du réseau cristallin de l’anode
lorsque les atomes de Cu et Fe sont oxydés se rassemblent en cristaux qui tombent au fond de la
cuve.
On obtient ainsi à la cathode du cuivre pur sans Fer ni Argent.
3) La réaction électrochimique à la cathode est Cu
2+
+ 2
e
–
= Cu
(S)
.
Pendant une durée
τ
où circule une intensité
I
constante, il passe la charge
q
=
Iτ
, soit une
quantité de matière d’électrons
A
q
n
N e
=
I
τ
=
F
. D’après le processus écrit ci-dessus, la quantité de
cuivre formée est
Cu
2 2
n I
n
τ
= =
F
si le rendement faradique est unitaire. La masse de cuivre obtenue
est donc
Cu Cu
2
I
m M
τ
=
F
. En prenant
m
Cu
= 0 en
t
= 0, on peut écrire
Cu
Cu
( )
2
IM
m t t
=
F
.
Le courant étant constant, la puissance reçue par l’électrolyseur est P =
∆U
.
I
et l’énergie
consommé entre
t
= 0 et
t
est
W
(
t
) =
∆U
.
I.t
soit
Cu
Cu
2 ( )
( )
m t
W t U M
= ∆
F
.
i
∆
U
U
Cu
(S)
Cu
2+
Cu
(S)
Cu
2+
H
2(G)
H
2
O
i
C
= – i
A
i
A
Cu
(S)
Cu
2+
Cu
(S)
Cu
2+
Ag(
S)
Ag
+
Ag
(S
Ag
+
Fe
(S)
Fe
2+
Fe
(S)
Fe
2+
0,8
0,34
–0,44
i
U (en V)
 6
6
1
/
6
100%