Syndrome du QT court

Revue
Syndrome du QT court
Fabrice Extramiana
Service de cardiologie, hôpital Lariboisière, 2 rue Ambroise-Paré, 75475 Paris Cedex 10
Résumé.Le syndrome du QT court est un syndrome génétique récemment découvert, responsable de mort subite, caractérisé par
l’association d’un intervalle QT court (QTc < 320 ms) sur l’électrocardiogramme et de la survenue de troubles du rythme auriculaire et/ou
ventriculaire sur un cœur par ailleurs considéré comme sain. Cinq ans après la première description clinique, il existe déjà trois formes décrites,
correspondant à des mutations dans trois gènes différents et aboutissant à une augmentation des courants ioniques repolarisants IKr, IKs et IK1.
Certaines études suggèrent que la majoration de la dispersion transmurale de la repolarisation consécutive à un raccourcissement hétérogène
de la durée de la repolarisation ventriculaire pourrait constituer le substrat arythmogène de ce syndrome. L’implantation d’un défibrillateur
automatique semble actuellement le meilleur traitement préventif chez les patients symptomatiques présentant des antécédents familiaux de
mort subite. Cependant, l’histoire naturelle de ce syndrome est encore mal connue, et s’il est important d’en faire le diagnostic, la prise en charge
en reste encore difficile.
Mots clés : QT court, mort subite, canalopathie
Abstract. Short QT syndrome. The short QT syndrome is a novel cause for sudden cardiac death. It is characterized by a short QT interval
(QTc < 320 ms) in the ECG, the absence of structural heart disease, a familial history of sudden cardiac death and major (resuscitated cardiac
arrest, syncope) or minor (palpitations, dizziness, atrial fibrillation) arrhythmic events. Five years after the first description, mutations in genes
encoding for 3 different potassium currents (IKr, IKs and IK1) have been associated with the syndrome. Experimental data suggest that
heterogeneous abbreviation of action potential duration among the different cell types spanning the ventricular wall creates the substrate for the
genesis of ventricular arrhythmias under conditions associated with short QT intervals. So far, the implantation of a defibrillator appears to be
the safest choice in symptomatic patients with a family history of sudden cardiac death. However, the natural history of the syndrome is not yet
well documented making the treatment decision process difficult. Nevertheless, this new syndrome deserves to be recognized in order to
prevent sudden cardiac death.
Key words: short QT syndrome, sudden cardiac death, potassium currents
Le syndrome du QT court est le
dernier né des syndromes généti-
ques responsables de mort subite.
Alors que l’association entre un QT
court et le risque de mort subite avait
été suspectée par des études épidé-
miologiques [1], l’identification du
syndrome est toute récente, avec une
première publication datant de l’an-
née 2000 [2]. En quelques années seu-
lement, une cause génétique a pu être
démontrée et nous connaissons déjà
trois types génétiques de ce syndrome.
Cette rapidité des découvertes aboutit
à une situation inédite dans laquelle la
génétique du syndrome est mieux
connue que son histoire naturelle.
D’autre part, le nombre d’articles pu-
bliés sur ce syndrome est en passe de
dépasser le nombre connu de patients
qui en sont atteints.
Il est donc très important d’avoir à
l’esprit que les données concernant ce
syndrome et que nous nous proposons
de passer en revue dans cet article
doivent être considérées comme très
préliminaires et susceptibles de modi-
fications rapides.
Définition et description
du syndrome
Le syndrome du QT court est ca-
ractérisé par l’association d’un inter-
valle QT court sur l’électrocardio-
gramme et de la survenue de troubles
du rythme auriculaire et/ou ventricu-
laire sur un cœur par ailleurs consi-
déré comme sain [2-4].
m
t
c
Correspondance : F. Extramiana
mt cardio 2005 ; 1 : 475-8
mt cardio, vol. 1, n° 5, septembre-octobre 2005 475
Revue
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

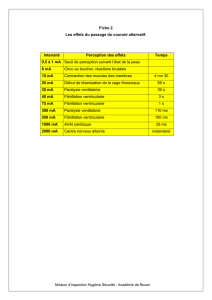
Description électrocardiographique
La notion de QT court doit être quantifiée et si l’on
retient un QTc < 320 ms, cette limite est pour l’heure
encore empirique et sujette à caution. En effet, l’intervalle
QT dans ce syndrome est caractérisé par un défaut d’adap-
tation à la fréquence cardiaque [5]. Il en résulte que
l’utilisation des formules classiques de correction de l’in-
tervalle QT en fonction de la fréquence cardiaque (par
exemple la formule de Bazett) est probablement inadaptée
dans ce syndrome. En l’absence de données plus précises
pour l’instant, il est conseillé de mesurer l’intervalle QT à
des fréquences cardiaques pas trop rapides (< 80 bpm).
Les cas publiés jusqu’à présent montrent des intervalles
QT dont le caractère « très court » est évident (figure 1).Il
n’est cependant pas exclu que ces cas correspondent à des
formes caricaturales et que des intervalles QT moins net-
tement courts puissent être associés à des événements
rythmiques [6]. Dans la plupart des cas publiés, le rac-
courcissement de l’intervalle QT semble associé à une
onde T symétrique, pointue et positive débutant juste
après la fin du QRS [4, 7, 8]. Il semble cependant que ce
caractère symétrique de l’onde T ne soit pas trouvé dans
toutes les formes du syndrome [9].
Description clinique
Sur le plan clinique, les patients atteints du syndrome
ont par définition un cœur normal. Sur le plan fonctionnel,
les patients peuvent être asymptomatiques, présenter des
palpitations, des malaises, des syncopes ou des morts
subites. Les palpitations et les malaises ont pu être associés
à la survenue d’épisodes de fibrillation auriculaire pa-
roxystique ou permanente [2]. D’autre part, les mémoires
des défibrillateurs ont permis de documenter que synco-
pes et morts subites étaient la conséquence d’arythmies
ventriculaires rapides (fibrillation ventriculaire et tachy-
cardie ventriculaire polymorphe) [8-10].
Les troubles du rythme peuvent survenir aussi bien
chez le nouveau-né qu’après 60 ans. La survenue d’une
fibrillation auriculaire peut précéder celle d’une syncope
et d’une mort subite. Cependant, la mort subite peut
également être le premier symptôme de la maladie.
Étiologie et mécanismes
Le syndrome peut se présenter sous la forme de cas
sporadiques, mais la notion de formes familiales a été
décrite très tôt, orientant vers une cause génétiquement
transmissible. Sur le plan théorique, le raccourcissement
de la durée de la repolarisation peut-être la conséquence
soit d’une augmentation de courants repolarisants, soit
d’une diminution de courants dépolarisants. Ce rationnel
a permis de cibler très vite des gènes candidats, et moins
de 5 ans après la première description clinique, nous
connaissons déjà trois formes correspondant à des
I
II
III
IVR
IVL
IVF
V1
V2
V3
V4
V5
V6
Figure 1.Fréquence cardiaque 52 bpm, QT = 280 ms
(d’après [4]).
Syndrome du QT court
mt cardio, vol. 1, n° 5, septembre-octobre 2005
476
Revue
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

mutations dans trois gènes différents (tableau 1). Les muta-
tions actuellement connues sont toutes à l’origine d’un
gain de fonction et donc d’une augmentation des courants
ioniques repolarisants. Ce gain de fonction peut être la
conséquence d’une augmentation de la conductance uni-
taire, d’une anomalie des propriétés de rectification, d’un
déplacement d’activation vers une phase plus précoce du
potentiel d’action ou encore d’une augmentation de cou-
rant repolarisant accélérant la phase terminale de la repo-
larisation [7-9]. Les gènes codants pour IK
ATP
et IK
Ach
sont
deux autres gènes candidats.
Peu de choses sont connues concernant les mécanis-
mes électrophysiologiques des arythmies observées dans
le syndrome du QT court. Les mutations décrites sont
toutes responsables d’un raccourcissement de la durée du
potentiel d’action ce qui, d’un point de vue théorique,
peut faciliter la survenue de fibrillation (au niveau auricu-
laire ou ventriculaire) par le biais d’une diminution de la
longueur d’ondes de circuits de réentrée. D’autre part, les
canaux ioniques n’étant pas répartis de manière homo-
gène dans le myocarde, l’anomalie d’un type de canal
peut aboutir à une majoration des hétérogénéités spatiales
de la repolarisation ventriculaire et constituer ainsi un
substrat fonctionnel de réentrée [11, 12].
Diagnostic
Compte tenu du faible nombre de patients connus
présentant le syndrome (quelques dizaines !), du faible
recul et de l’absence d’études importantes, il est particu-
lièrement difficile de faire des recommandations précises
pour la prise en charge et le traitement des patients atteints
de ce syndrome. Ce qui suit relève donc plus du bon sens
que d’une attitude médicale fondée sur les preuves.
La première étape du diagnostic doit consister à élimi-
ner les causes transitoires ou acquises de raccourcisse-
ment de l’intervalle QT. La durée de la repolarisation peut
être raccourcie par une hyperkaliémie, une hypercalcé-
mie, une acidose, une ischémie myocardique aiguë, une
hyperthermie, un phéochromocytome ou un traitement
digitalique ; toutes ces causes doivent donc être recher-
chées. Nous avons discuté plus haut des difficultés poten-
tielles de correction de la durée de l’intervalle QT en
fonction de la fréquence cardiaque. Le caractère court de
l’intervalle QT sera plus facilement documenté par la
répétition des électrocardiogrammes, par un enregistre-
ment Holter ECG des 24 heures et par une épreuve d’ef-
fort. Ces deux derniers examens permettent également
d’objectiver l’anomalie d’adaptation de l’intervalle QT
aux changements de fréquence cardiaque. L’exploration
électrophysiologique endocavitaire permet de mesurer
des périodes réfractaires courtes tant auriculaire que ven-
triculaire et le plus souvent de déclencher une fibrillation
ventriculaire [4, 5]. Il est trop tôt pour savoir si la positivité
de la stimulation ventriculaire programmée a une valeur
prédictive d’événement rythmique ventriculaire. La ré-
ponse à cette question ne pourra être connue rapidement
que si cet examen est réalisé de manière systématique.
Enfin, la notion de cœur sain doit être validée par une
échographie cardiaque et il ne faut pas oublier de réaliser
une enquête génétique et familiale la plus exhaustive
possible.
Traitement
L’histoire naturelle du syndrome étant encore très mal
connue mais caractérisée par un taux élevé de mort subite
dans les familles décrites, et en l’absence d’éléments
prédictifs de ce risque de mort subite, il semble légitime de
proposer l’implantation d’un défibrillateur automatique
aux patients symptomatiques ayant des antécédents fami-
liaux de mort subite [4, 7, 10]. Il faut signaler que les
caractéristiques de l’onde T observées dans ce syndrome
peuvent être à l’origine d’une sur-dérection de l’onde T
aboutissant à des chocs inappropriés [13].
La légitimité de l’implantation d’un défibrillateur est
encore renforcée par certaines observations de l’effet des
antiarythmiques dans ce syndrome. En effet, dans le syn-
drome du QT court de type 1 dans lequel la mutation est
responsable d’une augmentation de la composante rapide
du courant potassique rectifiant retardé (IKr), le blocage
sélectif de ce courant par le sotalol est inefficace aussi bien
in vitro qu’in vivo [4, 7]. En revanche, la quinidine qui
bloque de nombreux courants ioniques impliqués dans le
potentiel d’action ventriculaire semble capable d’allonger
la durée du QT, de restaurer une fréquence dépendante du
QT et de négativer la stimulation ventriculaire program-
mée chez les patients porteurs du syndrome du QT court
de type 1 [5, 14]. Ce résultat, qui n’est pas sans rappeler ce
qui est observé dans le syndrome de Brugada avec cette
molécule [15], devra cependant être validé par des études
cliniques prospectives et randomisées. Il faut également
rappeler que ni la quinidine ni les autres antiarythmiques
n’ont été testés dans les syndromes de type 2 et 3, de
description plus récente.
Enfin, il n’y a pas pour l’instant de données fiables pour
orienter la prise en charge des arythmies atriales observées
dans ce syndrome.
Tableau 1.Formes génétiques du syndrome du QT court
Type Gène Courant
ionique
Conséquences
fonctionnelles
Référence
SQTS1 KCNH2 IKr Gain de
fonction
7
SQTS2 KCNQ1 IKs Gain de
fonction
8
SQTS3 KCNJ2 IK1 Gain de
fonction
9
SQTS = short QT syndrome.
mt cardio, vol. 1, n° 5, septembre-octobre 2005 477
Revue
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

Conclusion
Dans l’état actuel des connaissances, le syndrome du
QT court pose surtout des questions. La prévalence de ce
syndrome n’est pas connue, il n’y a pas de valeur seuil de
durée de QT formelle pour en établir le diagnostic, il existe
une hétérogénéité génétique déjà importante potentielle-
ment responsable de différences phénotypiques. En l’ab-
sence d’outils de stratification du risque rythmique, la
seule solution thérapeutique envisagée pour l’heure est
l’implantation d’un défibrillateur automatique, alors
même que cette attitude n’est pas validée.
L’élément le plus important est probablement de savoir
rechercher un QT court chez les survivants d’une mort
subite ressuscitée, chez ceux atteints de FA ou présentant
des palpitations ou des syncopes et de recueillir un maxi-
mum d’informations sur ce nouveau syndrome dont l’his-
toire naturelle reste en grande partie à écrire.
Références
1. Algra A, Tijssen JG, Roelandt JR, Pool J, Lubsen J. QT interval va-
riables from 24 hour electrocardiography and the two year risk of
sudden death. Br Heart J 1993 ; 70 : 43-8.
2. Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval :
a new clinical syndrome? Cardiology 2000 ; 94 : 99-102.
3. Gussak I, Brugada P, Brugada J, et al. ECG phenomenon of idiopa-
thic and paradoxical short QT intervals. Card Electrophysiol Rev
2002;6:49-53.
4. Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome : a fami-
lial cause of sudden death. Circulation 2003 ; 108 : 965-70.
5. Wolpert C, Schimpf R, Giustetto C, et al. Further insights into the
effect of quinidine in short QT syndrome caused by a mutation in
HERG. J Cardiovasc Electrophysiol 2005 ; 16 : 54-8.
6. Viskin S, Zeltser D, Ish-Shalom M, et al. Is idiopathic ventricular
fibrillation a short QT syndrome? Comparison of QT intervals of
patients with idiopathic ventricular fibrillation and healthy controls.
Heart Rhythm 2004;1:587-91.
7. Brugada R, Hong K, Dumaine R, et al. Sudden death associated
with short-QT syndrome linked to mutations in HERG. Circulation
2004 ; 109 : 30-5.
8. Bellocq C, Van Ginneken AC, Bezzina CR, et al. Mutation in the
KCNQ1 gene leading to the short QT-interval syndrome. Circulation
2004 ; 109 : 2394-7.
9. Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT
syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ
Res 2005 ; 96 : 800-7.
10. Schimpf R, Bauersfeld U, Gaita F, Wolpert C. Short QT syn-
drome : successful prevention of sudden cardiac death in an adoles-
cent by implantable cardioverter-defibrillator treatment for primary
prophylaxis. Heart Rhythm 2005;2:416-7.
11. Kuo CS, Munakata K, Reddy CP, Surawicz B. Characteristics and
possible mechanism of ventricular arrhythmia dependant on the
dispersion of action potentials durations. Circulation 1983 ; 67 :
1356-67.
12. Extramiana F, Antzelevitch C. Amplified transmural dispersion of
repolarization as the basis for arrhythmogenesis in a canine
ventricular-wedge model of short-QT syndrome. Circulation 2004 ;
110 : 3661-6.
13. Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT syn-
drome and implantable cardioverter defibrillator treatment : inherent
risk for inappropriate shock delivery. J Cardiovasc Electrophysiol
2003 ; 14 : 1273-7.
14. Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome : phar-
macological treatment. J Am Coll Cardiol 2004 ; 43 : 1494-9.
15. Hermida JS, Denjoy I, Clerc J, et al. Hydroquinidine therapy in
Brugada syndrome. J Am Coll Cardiol 2004 ; 43 : 1853-60.
Syndrome du QT court
mt cardio, vol. 1, n° 5, septembre-octobre 2005
478
Revue
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.
1
/
4
100%