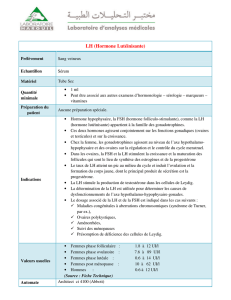
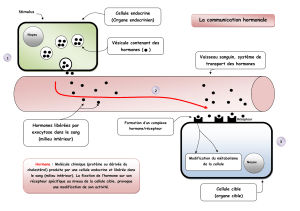
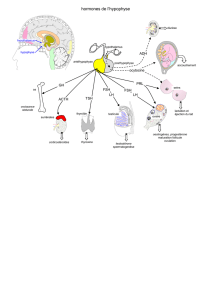
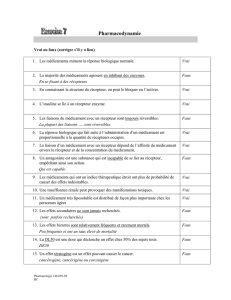
Maladies génétiques associées aux mutations - iPubli

S MINI-SYNTHÈSE
Maladies génétiques associées aux mutations
inactivatrices des récepteurs des gonadotrophines
C




 6
6
1
/
6
100%