Source électrospray : principes et applications en spectrométrie de masse

La source électrospray
La source électrospray a été crée en 1984 par Fenn
1
pour appliquer à des composés biologiques
d'intérêts médicaux, comme les pesticides, les acides nucléiques et la pénicilline. L'intérêt de
cette source est l'absence d'ion fragment des analystes dans le spectre de masse, et utilisation
simple rapide qui demande moins d'échantillon. En fait la fragmentation est plus ou moins
contrôlée dans la zone entre la sortie du capillaire, skimmer I et skimmer II. Cette source est
donc une source d'ionisation douce permettant facilement d'observer les ions précurseurs, ce qui
n'est pas le cas d'impact électronique
2
qui était largement utilisé à l'époque. De plus, certain
liaison fragile comme liaison non covalentes présentes dans la solution sont préservées dans la
phase gazeurs grâce à l'utilisation d'électrospray.
Depuis les chercheurs n'ont cessé d'améliorer
3
ce mode d'ionisation/désorption, selon les
molécules à analyser, la précision en masse requise. Des avancées ont aussi été faites avec le
couplage de la source à divers analyseurs (temp de vol TOF, quadripole, piège ionique,
spectromètre à transformée de Fourrier (FT/ICR, Orbitrap), mobilité ionique IMS). Les travaux
relatifs à cette source d'ions innovante se voient récompensés en 2002, lorsque Fenn reçoit le
Prix Nobel de Chimie.
1
Yamashita, M. and J.B. Fenn. 1984. Electrospray ion-source—another variation on the free-jet theme. J. Phys.
Chem. 88:4451-4459.
2
Edmond de Hoffman; Vincent Stroobant (2001). Mass Spectrometry: Principles and Applications (2nd ed. ed.).
John Wiley and Sons
3
Irina Manisali, David D.Y. Chen, Bradley B. Schneider Electrospray ionization source geometry for mass
spectrometry:
past, present, and future Trends in Analytical Chemistry, Vol. 25, No. 3, 2006

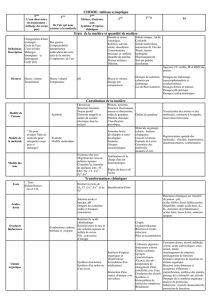
I - Parcours de l'échantillon de la source à l'analyseur
L'analyte, dilué dans un solvant adéquat, est injecté dans le capillaire métallique
d'introduction de la source ESI (Figure 1) au moyen d'une seringue, par exemple. Le débit utilisé
est de l'ordre de quelque µL/min à quelque centaines de µL/min et la température imposée au gaz
de nébulisation et évaporation se trouve en général entre 30 et 300°C. Ce gaz, en générale l'azote,
est introduit soit à contre courant, permettant d'augmenter le débit de l'échantillon (jusqu'à
plusieurs centaines de µL/min), soit à concourant.
Figure 1 : Schéma d'une source électrospray, de la zone de désolvatation, et de la zone de
transfert d'ions chargés
Sous la pression d'un gaz de nébulisation (N2) et éventuellement du gaz arrivant à contre
courant, la solution d'échantillon est vaporisée dans une zone à pression atmosphérique dans
laquelle règne un fort champ électrique. Celui-ci résulte d'une différence de potentiels (3 à 6kV)
appliquée entre la sortie du capillaire d'introduction et la contre électrode (figure ). C'est dans
cette zone que sont produits soit les ions solvatés après désorption, soit des agrégats chargés dont
l'état de charge, positif ou négatif, dépend du signe des potentiels appliqués.
Puis, ce nébulisât chargé atteint une zone de transfert matérialisée par un capillaire de
silice dont les extrémités sont métallisée. Ce dernier fait office d'interface entre la zone à
pression atmosphérique de la source et la zone suivant, qui se trouve à pression réduite. Des lors,
les ions subissent un gradient de pression : autour du torr en sortie du capillaire de transfert, au
Nébulisât
Contre électrode
N2(g)
N2(g)
Système pompage
SK1SK2
Hexapôles
Capillaire de transfert
Capillaire d’introduction
Nébulisât
Contre électrode
N2(g)
N2(g)
Système pompage
SK1SK2
Hexapôles
Capillaire de transfert
Capillaire d’introduction

dixième de torr pour la région de désolvatation et jusqu'à environ 10-6 torr pour la zone de
transfert des ions désolvatés.
Entre le capillaire de transfert et le premier cône (skimmer), une différence de potentiels
(ddp), de quelques dizaines de volts seulement, permet d'achever la désolvatation des ions
solvatés et des agrégats chargés. En effet, cette ddp accroit la vitesse des ions solvatés qui
subissent un grand nombre de collisions (mais selon un libre parcours moyen faible). Cela
signifie que les collisions sont peu énergétiques. Cette cascade de désolvatation faiblement
énergétique ne permet pas dans tous les cas de produire des ions totalement libres de solvant.
Celui-ci peut être éliminé dans une zone de pression plus réduite où les ions sont accélérés à
l'aide d'un second skimmer. Le libre parcours moyen des espèces est alors accru et les collisions
sont plus dures, éliminant ainsi les dernières molécules de solvant. Par contre une augmentation
trop forte de la ddp de cette région transfert suffisamment d'énergie interne aux espèces ionisées
pour produire la fragmentation de liaisons covalentes.
Enfin, les ions libres de solvant sont refocalisés au moyen de barres multipolaires afin
d'être éjectés de la région de désolvatation de la source vers l'analyseur. Cette géométrie est
couramment utilisée pour optimiser la transmission des ions.
II –Principes de l'ionisation/désorption par électronébulisation
II.1 – Processus d'ionisation/désorption
L'électronébulisation à pression atmosphérique repose sur une succession de phénomènes
physicochimiques. Leur description reste simple.
Tout d'abord, le champ d'électrique positifs (ou négatif) en sortie de capillaire
d'introduction provoque une accumulation de charges négatives (ou positives) à la surface du
liquide (Figure 2). Dans le cas des charges positives, le champ électrique permet de repousser les
charges négatives
4
, qui sont absorbées par les parois métalliques du capillaire d'introduction, qui
peut être à la terre. Ainsi se produit une réaction d'oxydation à l'interface liquide/capillaire tandis
qu'une réduction a lieu au niveau de la contre-électrode.
4
Arthur T. Blades, Michael G. Ikonomou, Paul Kebarle Mechanism of electrospray mass spectrometry. Electrospray
as an electrolysis cell Anal. Chem., 1991, 63 (19), pp 2109–2114

Figure 2 : Principe de l'ionisation/désorption par électronébulisation dans le cas d'une expérience
en mode d'ions positifs.
Sous l'effet des mouvements de ces charges, la surface de la solution se déforme jusqu'à
prendre la forme d'un cône (nommé cône de taylor). Dès que les pulsions coulombiennes entre
charges positives dépassent la tension de surface du liquide, un jet de gouttes chargées est émis à
partir de la pointe du cône. Leur diamètre est de l'ordre du micromètre. Il dépend de la valeur du
potentiel électrique, du débit de la solution, de la nature du solvant et de la température du gaz
N2.
L'optimisation de chacun de ces paramètres est primordiale pour permettre l'existence de
ces gouttes tout en évitant la modification des propriétés des ions et complexes préformés en
solution. Elles subissent alors une évaporation partielle du solvant, ce qui concentre les charges.
Les forces de répulsion coulombiennes sont toujours en compétition avec les forces de cohésion
qui maintiennent la gouttelette. A nouveau, une limite est atteinte (limite de Rayleigh), définit
par l'équation suivante.
oRayleigh rq
2/3
max
8
e-
Ddp = 3 à 6kV
Capillaire de
transfert
Echantillon liquide
Cône de taylor Contre électrode
Pression atmosphérique

qRayleigh : Charges moyenne de la goutte. γ: Tension de surface du solvant.
rmax : Rayon de la goulette. εo: Permittivité du vide.
A ce stade, au moins trois modèles de production des ions en phase gazeuse selon les
étapes suivantes sont proposés.
En 1968, Dole et al.
5
présentent un modèle connu sous le nom de théorie de la charge
résiduelle (Figure 3). Leur conception du processus de libération des ions en phase gazeuse se
base sur une continuté de la fission des gouttelettes en alternance avec l'évaporation du solvant.
D'après eux, l'évaporation totale du solvant, à partir de gouttelettes d'un rayon critique de 100 Ǻ,
mène à la naissance d'une molécule intacte dépourvu de solvant et chargée {macro-ion [M+nH]n+
dans le cas d'une ionisation positive, où M représente l'analyte}.
Figure 3 : Schéma relatif à la théorie de al charge résiduelle de Dole et al (S représente une
molécule solvant)
Plus tard en 1979, Iribarne et Thomson
6
développent le modèle de l'évaporation ionique à
partir de leurs études. Ce modèle repose sur l'émission, à partir d'une goulette chargée, d'un
agrégat formé d'un ion et de molécules de solvant [M+mM+nN]n+ (où S désigne une molécule de
solvant). L'émission d'un tel système est viable si on considère trois états (Figure 4): la
gouttelette chargée (état initial), et la gouttelette chargée de laquelle commence à ce détacher
l'agrégat [M+mS+nH]n+ (état de transition), et la gouttelette réduite séparée de l'agrégat.
L'enchainement de ces états est gouvernée par le nombre de charges portées par la gouttelette et
le rayon de celle-ci1. Iribarne et Thomson ont démontré que les paramètres critiques et le
5
Malcolm Dole, L. L. Mack, R. L. Hines, R. C. Mobley, L. D. Ferguson, and M. B. Alice Molecular Beams of Macroions
J. Chem. Phys. 49, 2240 (1968)
6
Iribarne, J. V.; Thomson, B. A. On the evaporation of small ions from charged droplets Journal of Chemical
Physics, Volume 64, Issue 6, pp. 2287-2294 (1976).
Evaporation
Du solvant
Fission des
gouttes
[M+mS+nH]n+ [M+nH]n+
Ions solvatés en
phase gazeuse
Ions en
phase gazeuse
 6
7
8
9
10
11
12
13
6
7
8
9
10
11
12
13
1
/
13
100%