octapharma - E-Compendium.be

Société pharmaceutique
(OCTAPHARMA)
1. DENOMINATION DU MEDICAMENT
GAMMANORM, 165 mg/ml, solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Immunoglobuline humaine normale (SC/IMIg)
Immunoglobuline humaine normale 165 mg/ml*
*Correspond à la teneur en protéines humaines, dont au moins 95% d’IgG.
Un flacon de 6 ml contient : 1 g * d’immunoglobuline humaine normale
Un flacon de 10 ml contient : 1.65 g * d’immunoglobuline humaine normale
Un flacon de 12 ml contient : 2 g * d’immunoglobuline humaine normale
Un flacon de 20 ml contient : 3.3 g * d’immunoglobuline humaine normale
Un flacon de 24 ml contient : 4 g * d’immunoglobuline humaine normale
Un flacon de 48 ml contient : 8 g * d’immunoglobuline humaine normale
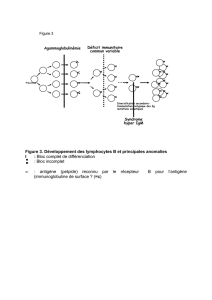
Répartition des sous-classes d’IgG :
IgG1 59%
IgG2 36%
IgG2 4.9%
IgG4 0.5%
IgA max. 82.5 microgrammes/ml
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Solution injectable
La préparation liquide est transparente ou légèrement opalescente et incolore ou jaune pâle ou marron clair.
4. DONNEES CLINIQUES
4.1 Indications thérapeutiques
Traitement de substitution des déficits immunitaires primaires chez les adultes et chez les enfants (de 0 à 18 ans), comme :
- les agammaglobulinémie et hypogammaglobulinémie congénitales
- le déficit immunitaire variable commun (DIVC)
- le déficit immunitaire combiné sévère
- les déficits en sous-classes d'IgG avec infections récurrentes
Traitement de substitution du myélome ou de la leucémie lymphatique chronique avec hypogammaglobulinémie secondaire sévère et
infections récurrentes.
4.2 Posologie et mode d’administration
Posologie
Traitement de substitution
Le traitement doit être initié et surveillé par un médecin expérimenté dans le traitement des déficits immunitaires.
Il peut être nécessaire d'adapter la posologie à chaque patient, en fonction des données pharmacocinétiques et de la réponse clinique.
Les posologies suivantes sont fournies à titre indicatif.
La dose administrée par voie sous-cutanée doit permettre d'atteindre une concentration stable en IgG. Une dose initiale d'au moins 0,2
à 0,5 g/kg peut être recommandée. Lorsque l’état d’équilibre en IgG est atteint, les doses d'entretien sont administrées à intervalles
réguliers pour atteindre une dose mensuelle cumulée de l’ordre de 0,4 à 0,8 g/kg.
Les concentrations minimales doivent être mesurées pour ajuster la posologie et l’intervalle d'administration.

Pour l'administration par voie intramusculaire, voir ci-dessous.
Patients en pédiatrie
Il existe des données chez les enfants (de 0 à 18 ans) souffrant de DIP.
Comme pour les adultes, les concentrations minimales devront être mesurées pour ajuster la posologie et l'intervalle d'administration.
Lorsque les concentrations en IgG ont atteint l'état d'équilibre, des doses d'entretien d'environ 80 à 100 mg/kg/semaine sont
habituellement administrées pour obtenir une dose mensuelle cumulée de l’ordre de 0,4 à 0,8 g/kg. Si un traitement à domicile est
envisagé, il est recommandé de consulter un médecin spécialisé dans le traitement des patients à domicile. Les parents du patient seront
formés à l’utilisation d’un guide-seringue, aux techniques de perfusion, à la tenue d’un journal de traitement et aux mesures à prendre
en cas d'effets indésirables graves.
Mode d’administration
Gammanorm doit être administré par voie sous-cutanée ou intramusculaire. Dans les cas particuliers où la voie sous-cutanée ne peut
être utilisée, de faibles doses de Gammanorm peuvent être administrées par voie intramusculaire.
La perfusion par voie sous-cutanée pour le traitement à domicile doit être mise en place par un médecin spécialisé dans le traitement
des patients à domicile. Le patient sera formé à l’utilisation d’un guide-seringue, aux techniques de perfusion, à la tenue d’un journal de
traitement et aux mesures à prendre en cas d'effets indésirables sévères.
Perfusion par voie sous-cutanée par pompe
La posologie habituelle est 0,6 ml (100 mg) de Gammanorm par kg de poids corporel une fois par semaine, pouvant être administrée
en plusieurs sites de perfusion. Vitesse de perfusion initiale : 10 ml/heure/pompe. La vitesse de perfusion peut être augmentée
progressivement, à raison de 1 ml/heure/pompe toutes les trois à quatre semaines. La dose maximale pouvant être administrée est 40
ml/heure en utilisant deux pompes simultanément.
Lors de la prescription de doses élevées, il est recommandé de les fractionner en plusieurs doses et de les administrer en différents
sites.
L’injection par voie intramusculaire doit être réalisée par un médecin ou par une infirmière.
4.3 Contre-indications
Hypersensibilité à la substance active ou l'un des excipients mentionnés à la rubrique 6.1.
Gammanorm ne doit pas être administré par voie intraveineuse.
Gammanorm ne peut être administré par voie intramusculaire en cas de thrombocytopénie sévère ou d'autres troubles de l’hémostase.
4.4 Mises en garde spéciales et précautions d’emploi
L’administration accidentelle de Gammanorm dans un vaisseau sanguin risque de provoquer un choc.
La vitesse de perfusion recommandée dans la rubrique 4.2 ("Mode d'administration") doit être scrupuleusement respectée.
Les patients doivent être suivis de près et maintenus sous observation étroite pendant la durée de la perfusion et au moins 20 minutes
après la perfusion, afin de s’assurer qu’aucun effet indésirable ne survienne.
Certains effets indésirables peuvent survenir plus fréquemment chez les patients traités par une immunoglobuline humaine normale pour
la première fois ou, dans de rares cas, lorsque les patients changent d'immunoglobuline humaine normale ou lorsque le traitement a été
interrompu pendant plus de huit semaines.
Des vraies réactions d'hypersensibilité sont rares. Elles peuvent se développer en particulier dans les cas très rares de déficit en IgA
avec anticorps anti-IgA ; ces patients doivent être traités avec précaution.
Très rarement, les immunoglobulines humaines normales peuvent provoquer une chute de la pression artérielle suivie d’une réaction
anaphylactique, même si les patients ont présenté une bonne tolérance à un traitement précédent par une immunoglobuline humaine
normale.
Les complications potentielles peuvent souvent être évitées en :
- s’assurant que les patients ne sont pas sensibles aux immunoglobulines humaines normales, en commençant par injecter lentement le
produit (voir rubrique 4.2).
- surveillant attentivement les patients pendant la période d’administration. Les patients recevant pour la première fois une
immunoglobuline humaine normale, les patients qui ont changé de spécialité ou pour qui une longue période s’est écoulée depuis la

dernière perfusion, doivent être plus particulièrement surveillés pendant la première perfusion et pendant la première heure suivant la
perfusion, afin de détecter d'éventuels effets indésirables. Les autres patients doivent rester sous surveillance au moins 20 minutes après
l’administration.
Si l’on suspecte des réactions de type allergique ou anaphylactique, arrêter immédiatement la perfusion. En cas de choc, administrer le
traitement médical standard.
Thromboembolie
Les manifestations thromboemboliques artérielles et veineuses, y compris infarctus du myocarde, thrombose veineuse profonde,
accident vasculaire cérébral et embolie pulmonaire, ont été reliées à l’utilisation d’immunoglobulines. La prudence s’impose chez les
patients présentant des facteurs de risque préexistants de manifestations thrombotiques (tels qu’âge avancé, hypertension, diabète
sucré et antécédents de maladies vasculaires ou épisodes thrombotiques, patients atteints de troubles thrombophiliques héréditaires,
patients ayant subi des immobilisations prolongées, patients présentant une grave hypovolémie, patients atteints de maladies augmentant
la viscosité sanguine). Les patients doivent être tenus informés des premiers symptômes des manifestations thromboemboliques, tels
que souffle court, douleur et gonflement d’un membre, déficits neurologiques focaux et douleurs dans la poitrine ; il faut également leur
conseiller de consulter leur médecin traitant dès l’apparition de symptômes. Les patients doivent être suffisamment hydratés avant
d’utiliser des immunoglobulines.
Les mesures habituelles de prévention du risque de transmission d’agents infectieux par les médicaments préparés à partir de sang ou
de plasma humain comprennent la sélection rigoureuse des donneurs, la recherche de marqueurs spécifiques d’infection sur chaque don
et sur les mélanges de plasma et l’inclusion dans le procédé de fabrication d'étapes efficaces pour l’élimination/inactivation virale.
Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission
d’agents infectieux, ne peut pas être totalement exclu. Ceci s’applique également à tous les virus émergents ou inconnus ou les autres
types d’agents infectieux, .
Les mesures prises sont considérées comme efficaces contre les virus enveloppés tels que le VIH, le VHB et le VHC.
Les measures prises peuvent être d’efficacité limitée vis-à-vis des virus non enveloppés comme le VHA et le parvovirus B19.
L'expérience clinique ne rapporte pas de transmission du virus de l'hépatite A ou du parvovirus B19 par les immunoglobulines
humaines normales et il est également supposé que la concentration en anticorps contribue de façon importante à la sécurité virale du
produit.
Pour avoir un lien entre le patient et le produit administré, il est fortement recommandé d'enregistrer le nom du produit et son numéro
de lot à chaque administration de Gammanorm.
Gammanorm ne protège pas contre l’hépatite A.
Ce médicament contient 4,35 mmol (ou 100 mg) de sodium par dose (40 ml). A prendre en compte chez les patients contrôlant leur
apport alimentaire en sodium.
Population pédiatrique
Il n’existe pas de précautions ni de mises en garde spécifiques ou supplémentaires pour la population pédiatrique.
4.5 Interaction avec d'autres médicaments et autres formes d'interactions
Vaccins constitués de virus vivants atténués
L’administration d’immunoglobuline peut réduire – pendant une période d'au moins 6 semaines et jusqu'à 3 mois – l’efficacité des
vaccins constitués de virus vivants atténués, comme les vaccins contre la rougeole, la rubéole, les oreillons et la varicelle. Après
administration de ce produit, il faut attendre un délai de 3 mois avant de vacciner les patients avec des vaccins constitués de virus
vivants atténués. Dans le cas de la rougeole, la diminution d'efficacité du vaccin peut persister jusqu’à 1 an. Un contrôle des anticorps
doit donc être réalisé chez les patients vaccinés contre la rougeole.
Interférence avec les tests sérologiques
Après administration d’immunoglobuline, l’augmentation transitoire de la concentration de divers anticorps transférés passivement dans
le sang des patients peut entraîner des résultats faussement positifs des tests sérologiques.
La transmission passive d’anticorps anti-érythrocytaires, par exemple A, B, D, peut interférer avec certains tests sérologiques
(numération des réticulocytes, haptoglobine et Test de Coombs).
Population pédiatrique
Aucune interaction spécifique ou supplémentaire n’a été observée dans la population pédiatrique.
4.6 Fertilité, grossesse et allaitement
Grossesse

L’innocuité de ce médicament au cours de la grossesse n’a pas été établie par des essais cliniques contrôlés et Gammanorm doit par
conséquent seulement être administré avec précaution chez la femme enceinte ou qui allaite. L’expérience clinique obtenue avec les
immunoglobulines laisse supposer que des effets indésirables ne sont pas attendus au cours de la grossesse et sur le fœtus et le
nouveau-né.
Allaitement
Les immunoglobines passent dans le lait maternel et peuvent contribuer à protéger le nouveau-né des pathogènes qui utilisent les
muqueuses comme point d’entrée.
Fertilité
L’expérience clinique obtenue avec les immunoglobulines laisse supposer que des effets indésirables sur la fertilité ne sont pas attendus.
4.7 Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Aucun effet n’a été observé sur l'aptitude à conduire et à utiliser des machines.
4.8 Effets indésirables
Les effets indésirables sont rares avec Gammanorm. En cas de réaction sévère, l’injection doit être interrompue, un traitement
approprié doit être initié.
Les effets indésirables suivants ont été observés avec Gammanorm :
Très fréquent (≥ 1/10); fréquent (≥ 1/100, <1/10); peu fréquent (≥ 1/1,000,<1/100); rare (≥ 1/10,000, <1/1,000); très rare
(<1/10,000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles)..
Classes de systèmes
d’organes
Fréquents Rares Très rares
Affections du système
immunitaire
Hypersensibilité Choc anaphylactique
Affections du système
nerveux
Maux de tête
Etourdissement
Affections vasculaires Hypotension événements
thromboemboliques*
Affections gastro-
intestinales
Nausées
Vomissements
Affections musculo-
squelettiques et
systémiques
Lombalgie
Arthralgie
Troubles généraux et
anomalies au site
d’administration
Réaction au site
d’injection
Fièvre
Frissons
Fatigue
*Terme MedDRA de plus bas niveau (LLT)
Population pédiatrique
Aucun effet indésirable spécifique n’est attendu pour la population pédiatrique.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue
du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national
de déclaration.
Belgique
Agence fédérale des médicaments et des produits de santé
Division Vigilance
EUROSTATION II
Place Victor Horta, 40/ 40
B-1060 Bruxelles
Site internet: www.afmps.be
e-mail: adversedrugreactions@fagg-afmps.be
Pour obtenir des informations sur la sécurité virale, voir rubrique 4.4.
4.9 Surdosage

Les conséquences d’un surdosage ne sont pas connues.
5. PROPRIETES PHARMACOLOGIQUES
5.1 Propriétés pharmacodynamiques
Classe pharmaco-thérapeutique : immunsérums et immunoglobulines, immunoglobulines, humaines normales, pour administration
extravasculaire.
Code ATC : J06BA01
L'immunoglobuline humaine normale contient principalement des immunoglobulines G (IgG) présentant un large spectre d’anticorps
contre divers agents infectieux.
L'immunoglobuline humaine normale contient des anticorps IgG présents dans la population normale. Ce produit est préparé à partir
d’un pool de plasma provenant d'au moins 1 000 dons. La distribution des sous-classes d’immunoglobuline G est similaire à celle
présente dans le plasma humain. Des doses adaptées de Gammanorm peuvent ramener à une valeur normale des taux anormalement
bas d’immunoglobuline G.
Population pédiatrique
Aucune étude spécifique n’a été réalisée avec Gammanorm dans la population pédiatrique.
5.2 Propriétés pharmacocinétiques
Lors de l'administration par voie sous-cutanée d’immunoglobuline humaine normale, le taux maximal dans la circulation du patient est
atteint en 4 à 6 jours.
Les données fournies par les études cliniques montrent qu'une concentration minimale de Gammanorm peut être maintenue par
l'administration d'une dose de 100 mg/kg par semaine.
Lors de l'administration par voie intramusculaire, l’immunoglobuline humaine normale a une biodisponibilité dans la circulation du
patient après un délai de2 à 3 jours.
Les IgG et les complexes IgG sont fractionnés dans les cellules du système réticulo-endothélial.
Population pédiatrique
Aucune étude n’a été réalisée avec Gammanorm dans la population pédiatrique.
5.3 Données de sécurité préclinique
Il n'existe aucune donnée significative.
6. DONNEES PHARMACEUTIQUES
6.1 Liste des excipients
Glycine, chlorure de sodium et acétate de sodium, polysorbate 80, eau pour préparations injectables.
6.2 Incompatibilités
En l’absence d’études de compatibilité, cette spécialité ne doit pas être mélangée à d’autres médicaments.
6.3 Durée de conservation
3 ans
Le produit doit être utilisé immédiatement après la première ouverture.
6.4 Précautions particulières de conservation
A conserver au réfrigérateur (2ºC – 8ºC). Ne pas congeler. Conserver le flacon dans l’emballage extérieur.
Pendant sa durée de conservation le produit peut être conservé jusqu’à 1 mois à une température ne dépassant pas 25 ºC sans être à
nouveau réfrigéré pendant cette période. Le produit doit être détruit s’il n’a pas été utilisé à l’issue de cette période.
6.5 Nature et contenu de l'emballage extérieur
Flacons de 6 ml, 10 ml, 12 ml, 20 ml, 24 ml ou 48 ml de solution, verre de type I avec un bouchon (caoutchouc de bromobutyle) –
boîte de 1, 10 ou 20.
Toutes les présentations peuvent ne pas être commercialisées.
6.6 Précautions particulières d’élimination et manipulation
 6
6
1
/
6
100%