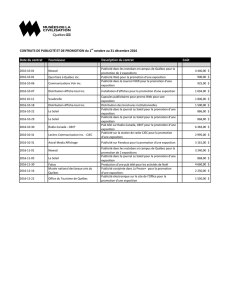
Nouvelles-éclair sur la santé

Nouvelles-éclair sur la santé
Bulletin trimestriel
Page 1 sur 12 | 3e TRIMESTRE DE 2014
Le Comité d’évaluation des médicaments examine les nouveaux médicaments
et les agents à surveiller au cours des réunions de juillet à septembre 2014
Le Comité d’évaluation des médicaments (CEM) d’Express Scripts Canada revoit chaque mois tous les avis de conformité
émis par Santé Canada pour les nouveaux médicaments. Le CEM s’assure ainsi de leur rôle thérapeutique et vérifie leurs
répercussions sur le secteur privé. Lesprix indiqués dans le présent document sont approximatifs et sont fournis uniquement
à titre d’information générale; ils ne sont pas destinés à servir de référence fiable aux fins de traitement des demandes de
règlement ni aux fins de remboursement des médicaments. Nous présentons ce bulletin trimestriel à nos clients à titre de
service à valeur ajoutée et espérons qu’ils le jugeront instructif, pertinent et utile.
NOUVEAUX MÉDICAMENTS
Firazyr® (acétate d’icatibant)
Présentation DIN et concentration Fabricant Classe AHFS
Solution pour injection
sous-cutanée
02425696 – 10 mg/ml (seringue
préremplie contenant 30 mg/3ml) Shire Orphan Therapies Inc. 92:32.00 – Inhibiteurs
du complément
Notes cliniques
L’angio-œdème héréditaire (AOH) est une maladie rare caractérisée par des épisodes récurrents d’angio-œdème nettement démarqués, sans
urticaire. Cette maladie affecte le plus souvent la peau ou les muqueuses des voies respiratoires supérieures et du tractus gastro-intestinal.
Sa prévalence n’est pas connue, mais on estime qu’elle se situe entre 1 personne sur 10 000 et 1 personne sur 150 000. Le nombre de cas
au Canada est estimé à environ 700. L’enflure disparaît par elle-même après deux à quatre jours en l’absence de traitement mais, par contre,
l’œdème du larynx peut provoquer une asphyxie mortelle et les douleurs causées par les crises gastro-intestinales peuvent être invalidantes.
En présence d’AOH, l’angio-œdème est attribuable à une production excessive de bradykinine, un puissant médiateur de la vasodilatation.
Des taux plasmatiques de bradykinine sept fois plus élevés que la normale ont été observés lors d’épisodes d’angio-œdème chez des patients
atteints d’AOH. L’histamine et les autres médiateurs mastocytaires n’entrent pas directement en jeu, ce qui explique l’absence de réponse aux
antihistaminiques et distingue cette forme d’angio-œdème de celle associée à l’urticaire.
Firazyr (icatibant) est un antagoniste synthétique du récepteur de la bradykinine B2. Ce polypeptide possède une structure analogue à celle de la
bradykinine et agit par antagonisme sélectif et compétitif des récepteurs de la bradykinine B2.
Place dans le traitement
Firazyr est indiqué pour le traitement des crises aiguës d’AOH chez l’adulte. Il s’agit du seul médicament approuvé que les patients peuvent
s’auto-administrer après avoir reçu l’enseignement nécessaire de la part d’un professionnel de la santé.
Indication(s)
Firazyr® est indiqué pour le traitement
des crises aiguës d’angio-œdème
héréditaire (AOH) chez l’adulte ayant un
déficit en inhibiteur de la C1 estérase.
Posologie
30 mg administrés par injection sous-cutanée
lente dans la région abdominale. Des doses
additionnelles peuvent être administrées à
intervalles de 6 heures, sans dépasser 3 doses
par période de 24 heures.
Autres options de traitement
Berinert® (inhibiteur de la C1 estérase
[humaine])

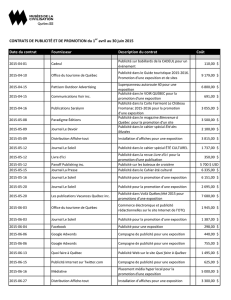
Nouvelles-éclair sur la santé
Bulletin trimestriel
Page 2 sur 12 | 3e TRIMESTRE DE 2014
Le Comité d’évaluation des médicaments examine les nouveaux médicaments
et les agents à surveiller au cours des réunions de juillet à septembre 2014
Prix comparatifs
Médicament Coût annuel estimatif
Firazyr®273 500 $
Berinert®prix non disponible
Répercussions / Suggestions pour la gestion des régimes
Répercussions moyennes. Nous recommandons l’autorisation préalable pour s’assurer que ce médicament est remboursé de façon appropriée.

Nouvelles-éclair sur la santé
Bulletin trimestriel
Page 3 sur 12 | 3e TRIMESTRE DE 2014
Le Comité d’évaluation des médicaments examine les nouveaux médicaments
et les agents à surveiller au cours des réunions de juillet à septembre 2014
Saflutan (tafluprost)
Présentation DIN et concentration Fabricant Classe AHFS
Solution ophtalmique 02425149 – 0,0015 % Merck Canada Inc. 52:40.28 – Médicaments O.R.L.O.,
analogues de prostaglandines
Notes cliniques
Saflutan (tafluprost) est un analogue fluoré de la prostaglandine F2α. L’acide de tafluprost, un analogue de prostaglandines, est un agoniste sélectif
du récepteur prostanoïde FP qui réduirait la pression intraoculaire en augmentant le débit uvéoscléral. Son mode d’action exact reste à déterminer.
Les analogues de prostaglandines sont légèrement plus efficaces que les bêta-bloquants non sélectifs. L’expérience clinique ne révèle aucun effet
indésirable systémique important avec les médicaments de cette classe thérapeutique, mais on a noté quelques effets oculaires comme des iris
de couleur brune devenus plus foncés, des cils allongés et une hyperémie conjonctivale légère. Ces agents peuvent tous être considérés comme
des traitements de première intention en raison de leur puissance et de leur excellent profil d’innocuité.
Si on opte d’emblée pour un traitement médical en présence d’un glaucome à angle ouvert, on envisage généralement l’emploi d’une
prostaglandine comme traitement de première intention. Les données semblent indiquer que les divers analogues de prostaglandines sont
d’efficacité semblable pour réduire la pression intraoculaire.
Place dans le traitement
Saflutan est le quatrième analogue de prostaglandines à faire son entrée sur le marché canadien. On peut s’attendre à ce qu’il occupe la même
place que d’autres analogues de prostaglandines comme traitement médical de première intention en cas de glaucome à angle ouvert.
Prix comparatifs
Indication(s)
*La version canadienne de la monographie du produit n’est
pas disponible; les renseignements sont tirés de l’information
posologique approuvée par la FDA pour Zioptan.*
Saflutan est indiqué pour réduire la pression intraoculaire
élevée chez les patients atteints de glaucome à angle ouvert
ou d’hypertension oculaire.
Posologie
Une goutte dans chaque œil
atteint, une fois par jour.
Autres options de traitement
XalatanMC (latanoprost)*; Lumigan® RC
(bimatoprost); Travatan® Z (travoprost)
*produits génériques offerts
Médicament Coût annuel estimatif
Saflutan prix non disponible
Apo®-Latanoprost 120 $
Lumigan® RC 430 $
Travatan® Z 365 $
Répercussions / Suggestions pour la gestion des régimes
Renseignements insuffisants

Nouvelles-éclair sur la santé
Bulletin trimestriel
Page 4 sur 12 | 3e TRIMESTRE DE 2014
Le Comité d’évaluation des médicaments examine les nouveaux médicaments
et les agents à surveiller au cours des réunions de juillet à septembre 2014
ElelysoMC (taliglucérase alpha)
Présentation DIN et concentration Fabricant Classe AHFS
Poudre pour injection intraveineuse 02425637 – 200 unités/fiole Pfizer Canada Inc. 44:00.00 – Enzymes
Notes cliniques
ElelysoMC (taliglucérase alpha), une enzyme hydrolytique et lysosomale administrée par perfusion intraveineuse qui cible spécifiquement le
glucocérébroside, est une forme recombinante active de la ß-glucocérébrosidase, une enzyme lysosomale exprimée dans des cellules de carottes
génétiquement modifiées mises en culture dans un bioréacteur jetable (ProCellEx®). La ß-glucocérébrosidase est une enzyme glycoprotéique
lysosomale qui catalyse l’hydrolyse du glucocérébroside (un glycolipide) en céramide et en glucose.
La maladie de Gaucher est une maladie de surcharge lysosomale dont il existe trois formes principales (types 1, 2 et 3) de même qu’une
forme fœtale et une autre forme avec atteinte cardiaque (maladie de Gaucher – ophtalmoplégie – calcification cardiovasculaire ou syndrome de
Gaucher). Sa prévalence est d’à peu près 1 personne sur 100 000. L’incidence annuelle de la maladie de Gaucher dans la population générale
est d’environ 1 personne sur 60 000, mais elle peut atteindre 1 personne sur 1 000 parmi les juifs ashkénazes. Les manifestations cliniques
de cette maladie varient grandement. La maladie de Gaucher de type 1 (90 % des cas) est la forme non neurologique chronique associée à une
splanchnomégalie (rate, foie), des anomalies osseuses (douleurs, ostéonécrose, fractures pathologiques) et une cytopénie. La maladie de Gaucher
de type 2, forme neurologique aiguë, caractérisée par une atteinte du tronc cérébral d’apparition précoce et d’évolution rapide, est associée à
une splanchnomégalie et entraîne le décès des patients avant l'âge de 2 ans. La maladie de Gaucher de type 3, forme neurologique subaiguë
qui atteint les enfants et les adolescents, se caractérise par une encéphalopathie évolutive (apraxie oculomotrice, épilepsie et ataxie) et par les
mêmes manifestations systémiques que la maladie de Gaucher de type 1.
Le déficit en glucocérébrosidase entraîne une accumulation de dépôts de glucosylcéramidase (ou bêta-glucocérébrosidase) dans les cellules du
système réticulo-endothélial du foie, de la rate et de la moelle osseuse (cellules de Gaucher).
Place dans le traitement
ElelysoMC offre aux patients atteints de la maladie de Gaucher une autre forme de traitement enzymatique substitutif en plus de celles déjà
offertes, soit Cerezyme® (imiglucérase) et VprivMC (vélaglucérase).
Indication(s)
ElelysoMC (taliglucérase alpha pour injection) est
indiqué pour le traitement enzymatique substitutif
de longue durée chez les patients ayant reçu un
diagnostic formel de maladie de Gaucher de type 1.
ElelysoMC peut aussi être utilisé chez les enfants
ayant reçu un diagnostic formel de maladie
de Gaucher de type 1, de même que pour le
traitement des manifestations hématologiques chez
les enfants ayant reçu un diagnostic formel de la
maladie de Gaucher de type 3.
Posologie
La dose doit être individualisée
pour chaque patient. ElelysoMC
doit être administré par perfusion
intraveineuse d’une à deux heures,
toutes les deux semaines. Les doses
initiales de taliglucérase alpha varient
entre 30 unités/kg et 60 unités/kg
de poids corporel, selon l’évaluation
clinique du médecin traitant.
Autres options de traitement
Cerezyme® (imiglucérase); VprivMC
(vélaglucérase alfa)

Nouvelles-éclair sur la santé
Bulletin trimestriel
Page 5 sur 12 | 3e TRIMESTRE DE 2014
Le Comité d’évaluation des médicaments examine les nouveaux médicaments
et les agents à surveiller au cours des réunions de juillet à septembre 2014
Prix
Médicaments Coût annuel estimatif*
ElelysoMC 187 000 $ à 373 000 $
Cerezyme®672 000 $
VprivMC 560 000 $
*supposant un poids corporel de 70 kg
Répercussions / Suggestions pour la gestion des régimes
Répercussions moyennes. Option potentiellement moins coûteuse que les traitements déjà offerts.
 6
7
8
9
10
11
12
6
7
8
9
10
11
12
1
/
12
100%