2016 - Pneumopathies interstitielles

Pneumologie 2016
Dr Gilles DARNEAU
PNEUMOPATHIES INFILTRATIVES DIFFUSES PID
Affections chroniques, non infectieuses et non malignes caractérisées par une association d’atteintes
isolées ou associées :
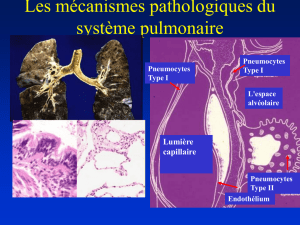
1. atteinte prédominante de l’interstitium pulmonaire, surface d'échange entre l'air (épithélium
alvéolaire) et le sang (endothélium capillaire), avec risque d’évolution vers une fibrose selon l’étiologie
= pneumopathie interstitielle diffuse avec parfois une composante alvéolaire,
2. atteinte des bronchioles respiratoires = bronchiolite,
3. atteinte du réseau vasculaire pulmonaire = angéite pulmonaire.
4. un risque évolutif commun : la fibrose pulmonaire et l’insuffisance respiratoire
Physiopathologie de l’atteinte pulmonaire interstitielle
Phase d’agression initiale = stade aigu (inflammation)
Phénomènes de destruction cellulaire, au niveau de l’endothélium capillaire et des cellules
alvéolaires,
Œdème lésionnel par trouble de perméabilité et exsudation dans la lumière alvéolaire d’un liquide
riche en protéines, entraînant la formation de membranes hyalines
L’interstitium est infiltré d’oedème et de cellules inflammatoires.
Phase de pneumonie interstitielle = stade chronique
L’oedème inflammatoire se résorbe
au niveau de l’épithélium une prolifération de pneumocytes II (« métaplasie cubique » de l’épithélium)
remplaçant les pneumocytes I
La lumière alvéolaire comporte, de même que l’interstitium, de nombreuses cellules : macrophages,
lymphocytes, polynucléaires granulomes par exemple.
Phase de fibrose
diminution de la cellularité des lésions et prédominance des dépôts de tissu conjonctif = fibrose
remplacement du tissu interstitiel par le collagène et prolifèration de fibroblastes
La fibrose détruit la structure d’échanges alvéolo-capillaires, rigidifie l’interstitium
Les bronchioles respiratoires sont souvent concernées par le processus fibrosant
A ce stade évolué de fibrose apparaissent des cavités kystiques (poumon en rayon de miel).
Principales étiologies
Environnement professionnel
- Pneumopathies d’hypersensibilité par inhalation d’antigènes organiques
poumon de fermier, éleveur d’oiseaux, des fromagers…
maladie des climatiseurs et humidificateurs.
- Pneumopathies interstitielles par inhalation de substances chimiques
Isocyanates par exemple
- Pneumoconioses :
Asbestose surtout, bérylliose, métaux durs, silicose exceptionnellement.
Environnement domestique (oiseaux, amiante …)
Médicaments :
Cytostatiques (bléomycine),
anti-infectieux (nitrofurantoïne),
cardiologiques (AMIODARONE
Maladies systémiques auto immunes
- Connectivite : lupus érythémateux disséminé, polyarthrite rhumatoïde, sclérodermie, syndrome de
Gougerot-Sjögren, myopathies inflammatoires
- Granulomatose : sarcoïdose, Wegener
- Angéite ou vascularite des vaisseaux de petit calibre
Facteur non connu (Fibrose Pulmonaire idiopathique) : 30% au moins

Pneumologie 2016
Dr Gilles DARNEAU
Un profil général élémentaire des PID
Il se caractérise par des :
- données cliniques : La dyspnée est le signe majeur, parfois s’accompagnant de toux ; les râles
crépitants fins à l’auscultation sont très évocateurs.
- données radiologiques : Les images réticulaires et nodulaires du « syndrome interstitiel »
radiologiques sont quasi constantes.
- données fonctionnelles respiratoires (EFR)
- trouble ventilatoire restrictif
- abaissement de la diffusion du CO,
- hypoxémie d’exercice puis de repos avec hypocapnie ;
Sarcoïdose
Affection multi systémique fréquente de cause inconnue touchant avec prédilection le poumon et le
système lymphatique et qui se caractérise par la formation de granulomes au niveau des organes
atteints.
Un facteur X entraîne une hyperproduction de lymphocytes au niveau des ganglions le plus souvent
thoracique et de l’interstitium pulmonaire. Ces lymphocytes s'organisent en granulomes au niveau des
organes qu'ils atteignent.
Maladie inflammatoire qui peut toucher tous les organes (peau, ganglions, foie, cœur, système
nerveux, yeux, rein...), et plus particulièrement le poumon : ganglions et interstitium.
Maladie souvent bénigne évoluant favorablement spontanément (80%), elle peut cependant évoluer
vers une fibrose pulmonaire en cas de PID.
Fréquente et souvent asymptomatique
Touche plus le sujet jeune, la femme, la race noire
FORME THORACIQUE :
Adénopathies médiastinales, symétriques, non compressives, asymptomatiques,
Plus rarement atteinte parenchymateuse avec risque d’évolution vers une fibrose pulmonaire pouvant
évoluer vers l'insuffisance respiratoire chronique.
Le diagnostic de certitude est parfois difficile à poser :
biopsie bronchique, pulmonaire ou ganglionnaire : granulome
lavage broncho-alvéolaire : lymphocytes en grand nombre
IDR tuberculine négative
FORMES EXTRATHORACIQUES :
peau : érythème noueux
yeux : iridocyclite
foie : hépatite
sys.nerveux : atteinte des nerfs périphériques
rein : insuffisance rénale
cœur : troubles du rythme parfois graves
TRAITEMENT :
Le plus souvent pas de TTT : évolution vers la guérison dans 80% des cas.
Corticothérapie dans les atteintes graves (rein, cœur, nerf) ou dans les atteintes pulmonaires avec
risque de fibrose évoluant vers l'IRC.
Fibrose pulmonaire idiopathique
Correspond à 30 % au moins des pneumopathies infiltratives diffuses, de survenue plus fréquente
chez le sujet âgé.
Signes révélateurs :
- toux et dyspnée puis hypoxie,
- aspect scanographique typique associant différents éléments :
- aspect d’atteinte micro nodulaire touchant plus volontiers les bases en zone périphérique,

Pneumologie 2016
Dr Gilles DARNEAU
- images de fibrose pulmonaire en rayons de miel à type de destruction du parenchyme pulmonaire
- images de bronchectasies correspondant en fait à la rétraction du parenchyme pulmonaire.
Bilan biologique
recherche de maladie systémique négative.
EFR
diminution des volumes mobilisables et en particulier de la CVF, diminution de la DLCO.
Gaz du sang
hypoxémie avec apparition secondaire d’une hypercapnie.
L’interrogatoire ne retrouve pas d’environnement domestique ou professionnel particulier ni
d’exposition médicamenteuse.
Un diagnostic peut être affirmé par une biopsie pulmonaire (chez les sujets jeunes uniquement).
Traitement
En cas de notion d’évolution de cette FPI, décision de mise sous ESBRIET (PIRFENIDONE) qui est le
seul agent anti fibrotique qui ait une action sur le déclin de la fonction respiratoire chez les adultes
atteints de FIP légère à modérée (CVF sup à 50 %, DLCO sup à 35 %).
Discussion chez le sujet jeune d’une greffe pulmonaire.
1
/
3
100%