Deficits immuns J-P Geslin

INSTITUT UNIVERSITAI
INSTITUT UNIVERSITAIINSTITUT UNIVERSITAI
INSTITUT UNIVERSITAIRE DE
RE DE RE DE
RE DE
FORMATION DES MAITRE
FORMATION DES MAITREFORMATION DES MAITRE
FORMATION DES MAITRES DE CRETEIL
S DE CRETEIL S DE CRETEIL
S DE CRETEIL
FACULTE DE BIOLOGIE
FACULTE DE BIOLOGIEFACULTE DE BIOLOGIE
FACULTE DE BIOLOGIE-
--
-
MEDECINE DE BOBIGNY
MEDECINE DE BOBIGNY MEDECINE DE BOBIGNY
MEDECINE DE BOBIGNY
1989- MARS 2000
LES DEFICITS IMMUNIT
LES DEFICITS IMMUNITLES DEFICITS IMMUNIT
LES DEFICITS IMMUNITAIRES
AIRES AIRES
AIRES
(A L’EXCEPTION DU SI
(A L’EXCEPTION DU SI(A L’EXCEPTION DU SI
(A L’EXCEPTION DU SIDA)
DA) DA)
DA)
INTRODUCTION : QUELQUES DEFINITIONS
I - LES DEFICITS IMMUNITAIRES PRIMITIFS :
A) LES DEFICITS SPECIFIQUES :
1) Déficits Immunitaires Combinés Sévères ou DICS
2)
Syndrome de Di George 3) Syndrome de Bruton
4) Le syndrome d’hyper IgM.
B) LES DEFICITS NON SPECIFIQUES :
1) Déficits génétiques constitutionnels en polynucléaires neutrophiles.
2) Déficits génétiques en complément.
II - LES DEFICITS
IMMUNITAIRES
ACQUIS :
A) LIES A DES
TRAITEMENTS
MEDICAMENTEUX.
B) LIES A DES
SYNDROMES
IMMUNO-
PROLIFERATIFS ET À
LEURS TRAITEMENTS
(à l’exclusion des formes
dues à des rétrovirus).
C) LES DEFICITS
LIES A DES
INFECTIONS :
D) LES DEFICITS
ASSOCIES A DES
TROUBLES
METABOLIQUES :
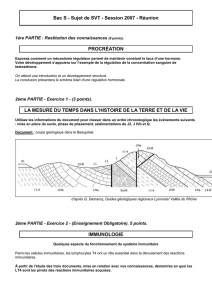
David, atteint d’un déficit immunitaire combiné sévère
(le syndrome des lymphocytes dénudés), dans sa bulle stérile.
JEAN
JEANJEAN
JEAN-
--
-PIERRE GESLIN
PIERRE GESLINPIERRE GESLIN
PIERRE GESLIN
PROFESSEUR AGREGE
PROFESSEUR AGREGEPROFESSEUR AGREGE
PROFESSEUR AGREGE

Jean-Pierre Geslin, professeur. 2
INTRODUCTION :
Les limites du sujet vont être définies par le sens qu’on donne au mot
« immunité » :
Initialement, le mot « immunité » était réservé à « l’acquisition par
l’organisme de propriétés de défense nouvelles et spécifiques à la suite
d’une infection ».
Par la suite, on a désigné sous ce nom l’ensemble des facteurs
humoraux et cellulaires (spécifiques ou non de l’agent agresseur) qui
protègent l’organisme contre les agressions infectieuses et parasitaires et
contre la prolifération maligne ou des tissus greffés.
Dans cette seconde acception, une déficience en un facteur humoral non
spécifique comme le lysosyme ou le complément ou une déficience en un type
cellulaire non spécifique comme les polynucléaires neutrophiles (qu’il s’agisse
d’une déficience qualitative ou quantitative) font partie intégrante du sujet.
1 enfant sur 100 naît avec une maladie génétique grave et on connaît plus
de 4000 maladies héréditaires.
La thérapie génique consiste à soigner les maladies héréditaires en
introduisant des gènes normaux dans l’organisme des patients, si possible à la
place exacte du gène déficient : on prélève des cellules au patients, on y
introduit le gène « thérapeutique » et on réintroduit les cellules manipulées
dans l’organisme. La thérapie génique peut être efficace même si toutes les
cellules ne sont pas « corrigées ».
Un certain nombre de déficits immunitaires primitifs sont susceptibles
d’être traités par cette méthode. On emploie des rétrovirus, comme vecteurs
des gènes thérapeutiques, rétrovirus capables d’infecter des types cellulaires
très variés, qui transforment leur ARN en ADN dans les cellules qu’ils
infectent et insèrent cet ADN dans les chromosomes. Les cellules cibles sont
ici, de préférence, les cellules de la moelle osseuse où se forme le sang.

Jean-Pierre Geslin, professeur. 3
On peut classer ces déficits d’après :
1) leur moment d’apparition,
2) leur nature,
3) ou leur cause.
1) Moment d’apparition du déficit :
a) Déficits primitifs
(= antérieur à la naissance). b) Déficits acquis
(= postérieur à la naissance).
DICS.
Di George.
Bruton.
Syndrome d’hyper IgM.
Déficits héréditaires en
polynucléaires neutrophiles
(P.N.).
Déficits en fractions du
complément.
SIDA.
Cytomégalovirus = C.M.V.
EBV.
Lèpre lépromateuse).
Hémopathies malignes (déficits variés).
2) Nature du déficit.
a) Déficit cellulaire
(diminution de l’activité et du nombre) : b) Déficits
humoraux :
des T4 (SIDA, CMV, thiopurines
: 6 mercaptopurine =
Purinéthol
et azathioprine = Imurel
).
des LB (
EBV, agents alkylants : cyclophosphamide =
Endoxan
et chlorambucil = Chloraminophène
).
des macrophages (polio).
des LB
, des LT et
des cellules phagocytaires :
corticoïdes.
Déficit en P.N. (granulomatose septique chronique,
maladie de Chediak-Higashi, médicaments ...).
des fractions
du complément.
des anticorps.
3) Cause du déficit.
a) Origine virale : HIV, CMV.
b) Origine bactérienne : lèpre.
c) Prolifération maligne :
* LLC diminution des immunités humorales et cellulaires.
* Kahler : IgG monoclonaux.
* Waldenström : IgM monoclonaux.
Hodgkin (un lymphome) :
de l’immunité cellulaire surtout.
d) Traitement immuno-suppresseur :
* Sérums antilymphocytaires.
* Substances immuno-suppressives.
e) Cause génétique :

Jean-Pierre Geslin, professeur. 4
I - LES DEFICITS IMMUNITAIRES PRIMITIFS :
A) LES DEFICITS SPECIFIQUES OU « ENFANTS - BULLE » (DICS,
SYNDROMES DE DI GEORGE ET DE BRUTON) :
A1) LES DICS (= déficits immunitaires combinés sévères) ou SCID ou DIMG
(= déficits immunitaires mixtes, graves) :
Déficits visibles dès l’âge de 2 mois après la naissance (avant, les nourrissons sont
partiellement protégés, de façon passive, par les anticorps d’origine maternelle).
Infections graves et répétées du fait de l’absence totale d’immunité :
•
••
•
virales (rougeole, varicelle, CMV = cytomégalo-
virus).
•
••
•
bactériennes (pyocyanique et colibacilles).
•
••
•
champignons (Candida albicans).
•
••
•
Protozoaires (Pneumocystis carinii).
⇒
⇒⇒
⇒ Outre les symptômes spécifiques, on peut noter :
des pneumopathies traînantes, des éruptions cutanées
récidivantes et des diarrhées.
⇒
⇒⇒
⇒ Les enfants
doivent être placés
dans une enceinte
stérile... une
« bulle » qui les
mettra à l’abri des
micro-organismes et
ceci jusqu'à
reconstitution
complète de leur
système
immunitaire : ce
sont les « Enfants
bulle » sinon, mort
généralement avant
6 ans (souvent plus
tôt), malgré les
traitements anti-
infectieux.
Thymus
atrophique,
ganglions et rate
déshabités.
Attention aux vaccination par
le BCG (vaccin bactérien
vivant) : risque de BCGite.
Antérieurement : risque avec
la vaccine ou cow-pox (vaccin
à virus vivant) avec risque de
« vaccine généralisée ».
Enfant atteint de DICS X et contraint de rester isolé en chambre
stérile en attente de greffe de moelle osseuse compatible ou de
thérapie génique.
Photo Florence Durand/Sipa press

Jean-Pierre Geslin, professeur. 5
Analyse :
•
••
•
absence de rosettes E
quand on met en présence
des globules rouges de
moutons à 4 ° Celsius et des
lymphocytes du sang humain
déficit en lymphocytes T
pas d’immunité à
médiation cellulaire.
•
••
•
des rosettes EAC (pour
Erythrocyte-Anticorps-Com-
plément ) qui permettent de
détecter certains lymphocytes B (mais tous les LB ne
portent pas ce récepteurs et les monocytes et
polynucléaires en sont pourvues)
ici,
LB.
•
••
•
une hypogammaglobulinémie franche (=
des
immunoglobulines) après disparition des anticorps
maternels.
Causes très variées mais maladies d’origine
génétique récessive en général, certaines liées au
chromosome X et d’autres autosomiques.
Chez certains DICS liés à l’X, un groupe de
récepteurs à diverses cytokines, normalement
présent à la surface des cellules précurseurs des lymphocytes T et des cellules
Natural Killer
dans la moelle osseuse et le thymus
« est inexistant ou bien inopérant ».
De ce fait, ces cellules (CD 34+) ne reçoivent plus les messages nécessaires à leur
différenciation. Plus précisément : une sous-unité de ces récepteurs est codée par le
gène gamma c = γ
γγ
γc et c’est cette sous-unité qui est commune aux récepteurs des
interleukines 2, 4, 7, 9 et 15.
Chez d’autres DICS liés à l’X, ce sont les lymphocytes B qui sont concernés.
Les DICS non liés à l’X peuvent correspondre à des déficits en enzymes ou à des
déficits en molécules de surface.
Qu’ils soient liés à l’X ou non, les DICS peuvent être combinés à d’autres
anomalies : c’est le cas :
Du Syndrome de Wiskott et
Aldrich ou WAS lié à l’X. Du syndrome d’Ataxie - Télangiectasie
héréditaire ou AT = syndrome de
Louis-Bar non lié à l’X.
ATTENTION : dans un cas sur 2, il est possible de détecter des lymphocytes T
circulants d'origine maternelle (microchimérisme).
↑
Schémas : Bach et Lesavre.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
1
/
24
100%