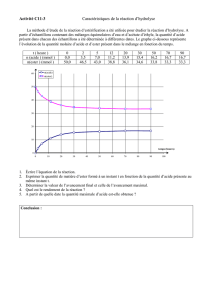
correction - Anne CURK

f
A
\
W
-o»
>
H
.
Ci»)
(T)
*J'&Àtfs>h*~à\f^
teo(**.oU
«0
^7Fx«
ou-he^r
w
10
r
roi
V
-"çk'
'«xrv»^
..
H
}]
CORRECTION PROBLEME COMPLET N°2

Le
site
électrophile
du DCC est
l'atome
de
carbone central possédant
une
charge partielle
+6

On
forme
:
t)s
A
-
^
HOBt
a un
rôle catalytique
: il
accélère
la
réaction tout
en
n'apparaissant
pas
dans
le
bilan.
II
ioue
le
rôle
d'assistant
nucléophile.
Première
voie
de
synthèse
BnO
£>
OBn
_
G
©NH
Bn°
\J
p
ICH-
}—{
BnO
OBn
OBn
OBn
7JH,
,*-
4-
C
-
II
s'agit d'une substitution nucléophile
:
Fhalogénoalcaiie
étant
primaire»
on
peut proposer
une
©
0
+ K, Br

On
représente
B'
par
H
^
.
H
H H
'CO2Et
CO2Et
Br
G
H©
R'-^f
^C02Et
H
H
H H
A/B
•CO2Et
^
-—
ÎO-H
prototropie
H©
•C02Et
d-H
H?N
<IO-H
'CO2Et
CQ2Ei
A/B
;o
C02Et
i«^C
JljL
rVA
C^UXAJlVAA
£>U,V
oL#
C
e^
4
"S
-ïV
"
-
.
"
\
^
•1
2
xoCi
^-^»
-wi
s^û/8
^
W|C
ft
k^
ff
*

14J
-
II
s'agit
de
formes
tautomères.
D
réagit d'abord avec
le DCC
pour donner
un
premier intermédiaire
D'
activé selon
un
mécanisme ana-
logue
à
celui
de la
question
5.
Puis:
BnO
BnO
^îouo
f*
Pour
les
deux autres
formes
tautomères,
en
considérant
une
catalyse acide (traces
de
H+
dans
le
mËieu)
:
f—v
-H
'o/
IH
.©
H
'O'
&Y
©
DH
- La
molécule
D
possède
un
centre
stéréogêiie
dont
la
configuration
est fixée. Ce
centre stéréogêne pro-
vient
de la
L-sérine (carbone
en a de la
fonction
carboxylate).
Le
couplage entre
D et F
passe
par la
formation
de
l'azlactone.
L'équilibre entre
les
trois
formes
tautomères
dont
deux possèdent
un
carbone
en a de la
fonction
ester
AX,}
entraine
une
racémisation
puisque lors
de la
protonation
qui
conduit
à la
première
forme,
deux configurations peuvent être obtenues
de
façon
équipro-
bable
selon
que
l'addition
se
fait
au
dessus
ou au
dessous
du
plan
de la
molécule
(mécanisme question
14),
 6
7
8
9
6
7
8
9
1
/
9
100%