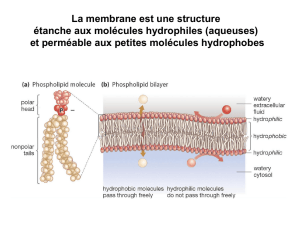
Matériel et méthodes Matériel et méthodes

Université de Reims Champagne Ardenne
Ecole Doctorale Sciences – Technologies – Santé
THESE
Présentée en vue de l’obtention du titre de
DOCTEUR DE L'UNIVERSITE
DE REIMS CHAMPAGNE ARDENNE
Spécialité : Biochimie et Biologie Cellulaire
Par
Gwenn PERROT
Membres du jury
Rapporteurs :
Dr. Emmanuelle LIAUDET-COOPMAN (Montpellier)
Pr. Philippe BOUCHER (Strasbourg)
Examinateurs :
Pr. Patrick HENRIET (Bruxelles)
Dr. Hervé EMONARD (Reims)
Directeurs de thèse :
Professeur Laurent MARTINY (Reims)
Docteur Stéphane DEDIEU (Reims)
Laboratoire SiRMa-Signalisation des Récepteurs Matriciels
Unité MEDyC, Matrice Extracellulaire et Dynamique Cellulaire
FRE CNRS/URCA N° 3481
UFR sciences Exactes et Naturelles de Reims
Université de Reims Champagne-Ardenne
IDENTIFICATION D’UN NOUVEAU PARTENAIRE DE LRP1
:
CD44, LE RECEPTEUR DE L’ACIDE HYALURONIQUE

Titre : Identification d’un nouveau partenaire de LRP1 : CD44, le
récepteur de l’acide hyaluronique
LRP1 est un récepteur d’endocytose multifonctionnel capable non seulement
d’interagir avec de nombreux ligands et de réguler leur endocytose, mais également de
moduler certaines voies de signalisation intracellulaire. Ce récepteur se révèle impliqué dans
de nombreux mécanismes physiologiques et pathologiques, comme l’athérosclérose, la
maladie d’Alzheimer ou encore le cancer. La multiplicité de ses partenaires fait émerger le
concept selon lequel LRP1 est capable de réguler le protéome membranaire et ainsi
d’influencer le comportement migratoire de nombreuses cellules. De plus, les résultats
récents obtenus au sein de notre laboratoire indiquent que ce récepteur d’endocytose est
capable de contrôler la dynamique d’adhérence des cellules tumorales. Nous avons donc
cherché à identifier un nouveau partenaire membranaire de LRP1, capable de participer à la
régulation de l’adhérence tumorale. Notre étude s’est portée sur le récepteur d’adhérence
CD44. L’utilisation de RAP, un antagoniste de LRP1 révèle que ce récepteur d’endocytose
module la présence de CD44 à la membrane plasmique. Nous avons également découvert que
ces deux récepteurs colocalisent fortement dans la cellule tumorale, que ce soit à la surface
cellulaire ou au niveau intracellulaire. De plus LRP1 et CD44 co-immunoprécipitent à partir
d’extraits totaux et membranaires, indiquant que ces deux récepteurs sont fortement associés
au sein d’un même complexe moléculaire. L’étude de la répartition membranaire du complexe
LRP1/CD44 a montré qu’il était en grande partie situé dans les radeaux lipidiques et
particulièrement les cavéoles. Toutefois ces structures n’apparaissent pas nécessaires à
l’établissement du complexe et ne sont pas impliquées dans les processus d’endocytose
relatifs à ces récepteurs. En effet, des expérimentations visant à quantifier l’endocytose de
CD44 couplées à différents traitements (β-MCD, conditions hyperosmotiques) révèlent que
l’endocytose de ce complexe implique principalement la voie des vésicules tapissées de
clathrine. Nous avons également démontré que ce mécanisme dynamique permettait de
réguler l’adhérence tumorale. Enfin, des travaux sur le shedding de CD44 ont permis
d’identifier les protéases impliquées (MT1MMP, ADAMs 10 et 17) et mettent en évidence un
effet protecteur de LRP1 sur le clivage de l’ectodomaine de CD44.
Mots clés : LRP1, CD44, endocytose, adhérence, cancer, radeaux lipidiques

e
ex
xÅ
Åx
xÜ
Üv
v|
|x
xÅ
Åx
xÇ
Çà
àá
á
Ce travail de thèse a été réalisé au sein de l’unité FRE CNRS/URCA N° 3481 MEDyC
et plus particulièrement au laboratoire SiRMa. Je tiens donc tout d’abord à remercier le
Professeur François-Xavier Maquart de m’avoir accueilli au sein de son unité.
Je tiens également à remercier le Professeur Philippe Boucher, ainsi que le Docteur
Emmanuelle Liaudet-Coopman d’avoir accepté d’être rapporteurs de cette thèse. De même, je
remercie le Professeur Patrick Henriet d’avoir apporté son jugement sur mon travail de thèse.
J’adresse également mes remerciements au Professeur Laurent Martiny et au Docteur
Stéphane Dedieu qui ont dirigé cette thèse. J’espère avoir été à la hauteur de vos attentes et je
vous remercie pour l’autonomie que vous m’avez accordée. Stéphane, votre passion pour la
recherche et votre optimisme m’ont aidée à avancer durant ces années, à ne pas baisser les
bras dans les moments de doutes et à mener au mieux mes travaux.
Je remercie également Hervé Emonard qui m’a prodigué de nombreux conseils. Un
grand merci également à Laetitia Parent et Cathy Hachet pour leur soutien technique et Hervé
Sartelet pour son aide dans l’isolement des radeaux lipidiques. Je remercie aussi Jérôme Devy
pour son aide dans le traitement de l’imagerie et Sébastien Almagro qui, même s’il n’est
arrivé qu’à la fin de mes travaux, m’a permis d’appréhender la vidéomicroscopie. Enfin,
merci à Fanja pour son aide en biologie moléculaire.
Je remercie également l’ensemble des membres de l’unité qui ont participé à ce travail
et tout particulièrement Christine Terryn et Hélène Bobichon pour leurs conseils et leur
participation à la mise au point des travaux d’imagerie.
Je tiens également à adresser mes remerciements aux personnes qui ont collaborées à
ce travail et notamment le Professeur Michel Khrestchatisky de la société VECT-HORUS
S.A.S., Marseille pour m’avoir permis d’utiliser les microdomaines de LRP1. Je remercie

également toutes les personnes participantes à l’ANR TIMPAD pour leur accueil à Marseille
et pour m’avoir fait découvrir la bouillabesse.
J’ai également une pensée pour les organismes ayant permis le financement de ce
travail et tout particulièrement le Ministère de l’Education Nationale et de l’Enseignement
Supérieur, le CIES, la région Champagne-Ardenne, le CNRS, l’ANR, et également la SFBBM
et la FEBS pour avoir en partie financer ma participation à des congrès nationaux et
internationaux.
Je tiens également à remercier les personnes m’ayant encadrée ou conseillée dans ma
fonction de moniteur, notamment Frédéric et Christelle (Oudot-)Delacoux, Marie-Pierre
Courageot, Gwenola Simon, Jean-Louis Druelle. Cette expérience m’a beaucoup appris et a
été très agréable à réaliser en leur compagnie.
J’ai bien sûr une pensée particulière pour Benoît Langlois qui m’a précédé. Je lui
souhaite un avenir plein de réussite et le remercie pour tous les conseils qu’il m’a fournis et la
passion pour la recherche qu’il transpirait. Je pense également à ceux que j’ai vu partir
comme Lucie, Bénito ou Farid, je leur souhaite beaucoup de bonheur personnel et
professionnel.
Ensuite, je voudrais exprimer toute la gratitude et l’amitié que j’ai envers tous les
membres du laboratoire SiRMa qui ont fait de ces quatres années passées avec eux une grande
expérience humaine. La joie et la bonne humeur que vous m’avez procurées m’ont permis de
toujours venir travailler avec le sourire même lorsque mes travaux devenaient pénibles. Je
pense particulièrement aux membres du bureau BI18 Marie-Line, Titof, Albin et Oliver, les
nombreux goûters que l’on a partagés resteront longtemps marqués sur mes hanches. Merci
aussi à la Coq, Jess, Callaghan, Dude, Nikétic, Sébastien, Nicolas, Stéphanie, Florence avec
qui j’ai passé plus que des bons moments et qui me manqueront terriblement. Nos
interminables conversations sur des sujets plus ou moins douteux, nos fous rires, je ne les
oublierai pas. J’ai bien sûr une pensée à tous les autres que je n’ai pas encore cité : Francine,
Aurélie, Emmanuelle, Vérène, Hassan, Laurent, Katia, Jean-Luc, Annie, Isabelle, Bertrand,
Stéphanie S., Michel et Manu.

J’ai aussi une pensée à tous les étudiants qui ont eu la chance de faire un passage au
sein du laboratoire. Certains sont restés, notamment Laurie à qui je souhaite le meilleur. Pour
ceux qui nous ont quittés mais sont restés dans la recherche, je leur souhaite bon courage ; je
pense surtout à Alexandra, Damien et Simon.
Enfin, je ne pouvais pas terminer ces remerciements sans un mot pour ma famille qui
m’a soutenu dans mes études. Votre fierté aide à gommer mon éternelle insatisfaction dans ce
que j’entreprends. Je pense bien sûr à mes parents, mes frères et sœurs et aussi à Marie-
Thérèse et Christophe qui sont passés par là également. Marie-Thé, la règle des 80/20 m’aide
maintenant à relativiser les choses.
Mes dernières paroles iront à Jérôme qui partage ma vie depuis bientôt 10 ans, il m’a
connu lorsque je n’étais même pas encore bachelière et m’aura accompagné jusqu’à ce que je
sois Docteur. Son soutien inconditionnel m’a aidé à tenir lorsque je voulais baisser les bras. Il
a toujours cru en moi et en ce que je faisais peut-être même plus que je n’y croyais moi-
même. Il m’a apporté énormément de bonheur et sans doute le plus beau cadeau que l’on
puisse faire à une femme : un petit garçon, notre Enaël.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
1
/
159
100%