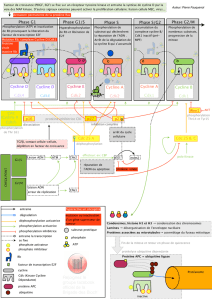
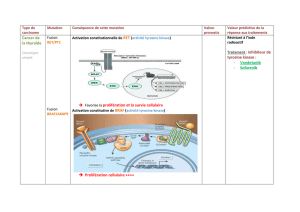
Caractérisation du rôle des protéines phosphatases impliquées

Caract´erisation du rˆole des prot´eines phosphatases
impliqu´ees dans la d´ephosphorylation de la prot´eine
kinase Greatwall lors de la sortie de mitose
Sheng Ma
To cite this version:
Sheng Ma. Caract´erisation du rˆole des prot´eines phosphatases impliqu´ees dans la
d´ephosphorylation de la prot´eine kinase Greatwall lors de la sortie de mitose. Biologie cel-
lulaire. Universit´e Montpellier, 2015. Fran¸cais. <NNT : 2015MONTS007>.<tel-01315940>
HAL Id: tel-01315940
https://tel.archives-ouvertes.fr/tel-01315940
Submitted on 13 May 2016
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-
entific research documents, whether they are pub-
lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destin´ee au d´epˆot et `a la diffusion de documents
scientifiques de niveau recherche, publi´es ou non,
´emanant des ´etablissements d’enseignement et de
recherche fran¸cais ou ´etrangers, des laboratoires
publics ou priv´es.

Délivré par L'Université de Montpellier
Préparée au sein de l’école doctorale CBS2
Et de l’unité de recherche UMR 5237
Centre de Recherches de Biochimie Macromoléculaire
Spécialité : Biologie Cellulaire et Moléculaire, Biochimie
Présentée par Sheng MA
Soutenue le 9 octobre 2015 devant le jury composé de
M. Olivier COUX, CRBM
Président
M. Jean-Pierre TASSAN, IGDR
Rapporteur
M. Lionel PINTARD, IJM
Rapporteur
Mme. Anne-Marie GENEVIERE, BIOM
Examinateur
Mme. Anna CASTRO, CRBM
Directrice de thèse
M. Thierry LORCA, CRBM
Co-Directeur de thèse
Caractérisation du rôle des protéines phosphatases
impliquées dans la déphosphorylation de la protéine kinase
Greatwall lors de la sortie de mitose

THESE DE DOCTORAT DE L'UNIVERSITE MONTPELLIER II
SCIENCES ET TECHNIQUES DU LANGUEDOC
Spécialité :
Biologie Cellulaire et Moléculaire, Biochimie
Ecole Doctorale :
Sciences Chimiques et Biologiques pour la Santé
Présenté par
Sheng MA
Le 9 octobre 2015
Pour obtenir le grade de
DOCTEUR DE L'UNIVERSITE MONTPELLIER II
Sujet de thèse :
Caractérisation du rôle des protéines phosphatases impliquées
dans la déphosphorylation de la protéine kinase Greatwall lors
de la sortie de mitose
Devant le jury composé de :
Dr. Olivier COUX
Président
Dr. Jean-Pierre TASSAN
Rapporteur
Dr. Lionel PINTARD
Rapporteur
Dr. Anne-Marie GENEVIERE
Examinateur
Dr. Anna CASTRO
Directeur de thèse
Dr. Thierry LORCA
Directeur de thèse

!
"!
« Entre le ciel et la terre, il n'y a qu'une demeure, temporaire. »
Bouddha, soutra.

!
#!
Remerciements
Tout d’abord, je voudrais remercier mes parents.
Et je voudrais remercier mes deux Maîtres : Liwu et Yongchao.
Merci à mon amour Dandan.
Merci à Adrien et Déborah.
Je remercie chaleureusement le Dr. Olivier COUX d’avoir accepté de présider mon jury de
thèse.
Je tiens à exprimer ma profonde gratitude aux Drs. Jean-Pierre TASSAN et Lionel PINTARD
pour m’avoir fait l’honneur de rapporter mon travail de thèse et au Dr. Anne-Marie
GENEVIERE pour avoir accepté de l’examiner.
Je voudrais remercier vraiment très chaleureusement mon co-directeur de thèse, M. Thierry
LORCA, un chercheur vraiment excellent ! Un très grand merci pour m’avoir accueilli dans
ton équipe, m’avoir fait découvrir le monde « phosphatase », m’avoir soutenu sans faillir et
surtout pour tout ce que tu as fait pour moi. Ton esprit de recherche, ta connaissance des
projets, ton attitude pour le travail, ta tolérance pour mes bêtises et ton « à fond, à fond », tout
cela demeurera en moi toute ma vie et me permettra d’avancer et de lutter dans les difficultés.
Je voudrais remercier mon co-directeur de thèse Mme. Anna CASTRO, un chercheur très
brillant. Un grand merci Anna pour tes conseils et ta disponibilité, ainsi que pour ta
stimulation. Ton expérience et ton efficacité aussi resteront en moi toute ma vie,
Merci Suzanne pour avoir partagé tes connaissances et pour tes conseils quotidiens. Tu as été
toujours très patiente devant mes questions et toujours prête à m’aider.
Merci Sophie, tu as vraiment joué un rôle important à des moments-clés. J’ai beaucoup
apprécié ton caractère, surtout ta franchise et la grande valeur des conseils que tu m’as
donnés.
Merci Coco, les discussions avec toi sont toujours riches et intéressantes.
Merci Khaled pour ton soutien quotidien, merci Julie pour tes conseils, merci Perle pour tes
informations, merci Lena pour ta vitalité et ta bonne humeur.
Je voudrais remercier M. Claude ALBAN : merci pour ton soutien pendant mon Master 2 et
pendant ma thèse. J’ai appris tellement de choses de ta part.
Je voudrais te remercier Jacques PIETTE, pour ta compréhension, ton soutien, ta gentillesse
et surtout pour ton aide.
Merci Claude DELSERT pour tes conseils pratiques et ton soutien.
Merci à toi aussi Gabriel pour ton appui et ton aide.
Merci Marion, François, Thom, Barbara, Damien P. et Ilaria, pour toutes vos bonnes idées et
votre aide.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
1
/
126
100%